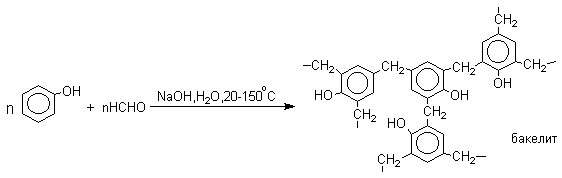
3.5.7. Конденсация фенолов с альдегидами и кетонами
Фенолы реагируют с формальдегидом в водном растворе в присутствии основания с образованием полимерного продукта, получившего название феноло-формальдегидной смолы, карболита или бакелита. В 1909 году Л.Бакелунд запатентовал способ получения этого первого синтетического высокомолекулярного соединения, которое сразу же нашло широкое применение в различных областях машиностроения, электротехники и быта, например, при изготовлении корпусов телефонов, электрических выключателей и т.д.
Взаимодействие феноксид-иона с формальдегидом напоминает альдегидную конденсацию с той лишь разницей, что роль нуклеофильного агента вместо енолят-иона выполняет амбидентный феноксид-ион, а карбонильной компонентой является формальдегид.
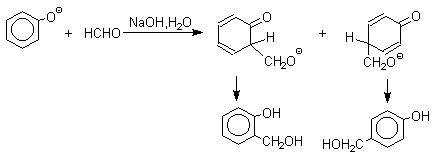
Подобно альдолям, орто- и пара-изомеры гидроксиметилфенола подвергаются дегидратации с образованием хинонметидов - соединений, родственных орто- и пара-хинонам.
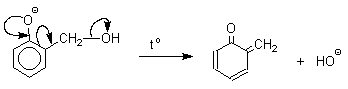
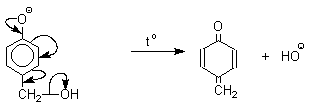
Последующее присоединение феноксид-иона к
хинонметиду представляет собой присоединение
амбидентного аниона к  ,
,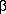 -непредельному кетону по Михаэлю.
-непредельному кетону по Михаэлю.
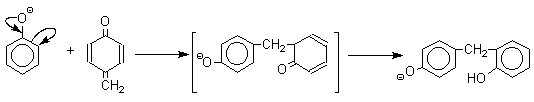
В результате дальнейшей поликонденсации в орто- и пара-положение к гидрокси-группе фенола получается трехмерная структура конечного продукта - бакелита. Бакелит представляет собой прозрачную смолу, где линейные звенья связаны "поперечными" связями в пара-положениях.
Фенол конденсируется с ацетоном в кислой среде с образованием так называемого бисфенола А.
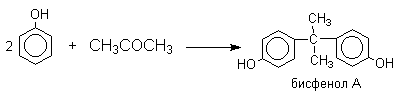
Получено много подобных продуктов конденсации фенолов с кетонами. Они находят применение в качестве антиоксидантов и мономеров для получения эпоксидных смол, получаемых, например, при конденсации бисфенола А с эпихлоргидрином:
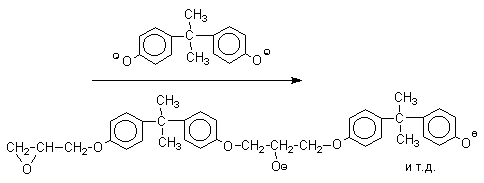
Эпоксидные смолы широко применяется в современной технике и в быту.
3.5.8. Карбоксилирование феноксид-ионов - реакция Кольбе
Оригинальный метод введения карбоксильной группы в ароматическое кольцо был открыт Г.Кольбе в 1860 году. При нагревании сухих фенолятов натрия или лития с СО2 при 150-180оС и давлении 5 атм, образуются натриевые или литиевые соли салициловой кислоты. В аналогичных условиях из фенолятов калия, рубидия и цезия получаются только соли пара-гидроксибензойной кислоты.
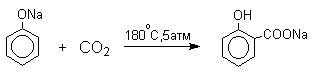
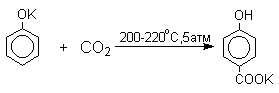
Такое различие в направлении карбоксилирования Na- и К-солей фенола принято объяснять различием в хелатообразовании этих двух катионов с атомом кислорода CO2 в переходном состоянии реакции приводящем к салициловой кислоте. Катионы натрия и, особенно, лития значительно более эффективны по сравнению с катионом калия в способности к образованию координационной связи с атомом кислорода.
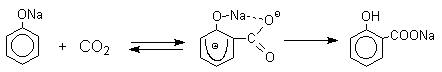
Предполагается, что для фенолятов калия, рубидия и цезия электрофильная атака осуществляется исключительно в пара-положение без какой-либо координации катиона по атому кислорода. Эта очень распространенная точка зрения на механизм реакции Кольбе все-таки не объясняет исчерпывающим образом диаметрально противоположное поведение фенолятов натрия и калия в этой реакции, так как известно, что катион калия способен к образованию комплексов с кислородными лигандами, например, краун-полиэфирами. Более подробное изучение указывает на то, что механизм этой внешне очень простой реакции намного более сложен, чем это принято считать.
Безводные моносалицилаты калия и рубидия при нагревании до 200-220оС дают ди-К- и ди-Rb-соли пара-гидроксибензойной кислоты и фенол.
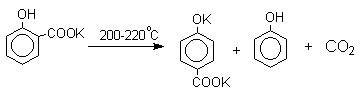
Дикалиевая соль салициловой кислоты перегруппировывается в дикалиевую соль пара-гидроксибензойной кислоты:
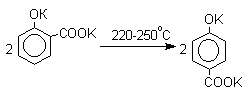
Натриевая соль пара-гидроксибензойной кислоты при нагревании превращается в динатриевую соль салициловой кислоты:
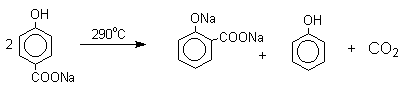
Из этого следует, что карбоксилирование щелочных фенолятов представляет собой обратимую реакцию и направление ее зависит только от природы катиона. В отличие от одноатомных фенолов двухатомные и трехатомные фенолы карбоксилируются в более мягких условиях. Так, резорцин карбоксилируется при пропускании СО2 в водный раствор его дикалиевой соли при 50оС. При этом образуется 2,4-дигидроксибензойная кислота.
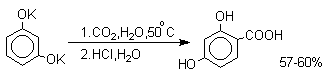
3.5.9. Азосочетание
Фенолы при взаимодействии с солями арендиазония в слабощелочной среде образуют арилазофенолы. Эта реакция получила название азосочетания. По своему механизму реакция азосочетания является реакцией электрофильного замещения, в которой соли арендиазония выступают в качестве электрофильных реагентов. В качестве примера приведем получение красителя пунцового 2R, который получается при азосочетании диазотированного мета-ксилидина и R-кислоты.
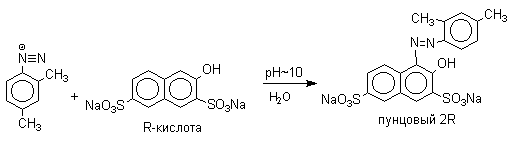
Подробно о реакции азосочетания можно прочитать в методической разработке О.А.Птициной "Ароматические диазосоединения" (1989г)
3.6. Перегруппировка Кляйзена аллилариловых эфиров
В 1912 году Л.Кляйзен открыл интересную и своеобразную перегруппировку аллиловых эфиров фенолов в аллилфенолы, которая стала прототипом для многих родственных сигматропных перегруппировок. Аллиловый эфир фенола при нагревании до 200-220оС превращается в орто-аллилфенол, т.е. аллильная группа мигрирует в орто-положение бензольного кольца.
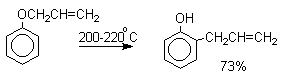
Если оба орто-положения заняты заместителями, то аллильная группа перемещается в пара-положение:
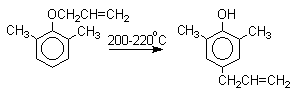
Установлено, что и орто- и пара-перегруппировки
являются внутримолекулярными реакциями первого
порядка, которые сопровождаются инверсией
мигрирующей аллильной группы, т.е. аллильная
группа присоединяется к бензольному кольцу
своим  -углеродным атомом.
-углеродным атомом.
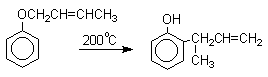
Из этого следует, что переходное состояние
перегруппировки Кляйзена должно быть
циклическим шестизвенным. Такое переходное
состояние включает шесть  -электронов
и является ароматическим, что составляет
движущую силу этой термической перегруппировки.
На последней стадии происходит изомеризация
циклогексадиенона в о-аллилфенол. Эта стадия
полностью аналогична изомеризации кетона в
енольную форму.
-электронов
и является ароматическим, что составляет
движущую силу этой термической перегруппировки.
На последней стадии происходит изомеризация
циклогексадиенона в о-аллилфенол. Эта стадия
полностью аналогична изомеризации кетона в
енольную форму.
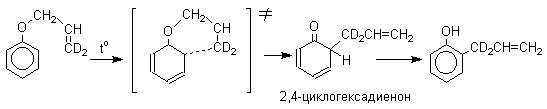
2,4-Циклогексадиенон является интермедиатом перегруппировки аллилариловых эфиров. Такой интермедиат может быть выделен при перегруппировке аллилового эфира 2,6-диметилфенола, когда аллильная группа мигрирует в пара-положение, поскольку енолизация кетона в фенол в этом случае не может происходить из орто-положения. Конечным результатом двух последовательных миграций аллильной группы является сохранение структуры мигрирующей группы.
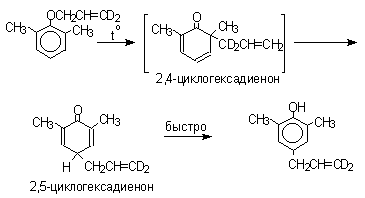
Кроме того, при проведении перегруппировки в присутствии малеинового ангидрида 2,4-циклогексадиенон улавливается в виде аддукта диенового синтеза.
Миграция аллильной группы характерна не только
для аллиловых эфиров фенолов. Аллиловые эфиры
енолов также подвергаются аллильной
перегруппировке. Например, аллилвиниловые эфиры
в результате миграции аллильной группы
превращаются в ,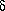 -ненасыщенные карбонильные
соединения.
-ненасыщенные карбонильные
соединения.
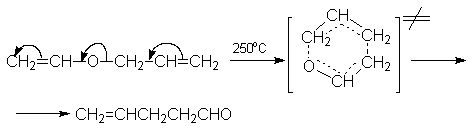
3.7. Окисление фенолов
Окисление пространственно незатрудненных фенолов относится к числу сложных, многостадийных процессов, механизм которых мало изучен. Очевидно лишь то, что механизм окисления может сильно меняться в зависимости от природы одно- или двухэлектронного окислителя. Сам фенол при окислении двухэлектронным окислителем - бихроматом натрия или MnO2 в серной кислоте образует с удовлетворительным выходом пара-хинон.
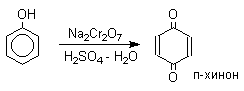
Универсальным одноэлектронным окислителем фенолов является соль Фреми - нитрозодисульфонат калия - редкий пример стабильного неорганического нитроксильного свободного радикала, полученного впервые еще в 1845 году. Окисление фенолов солью Фреми идет в очень мягких условиях по радикальному механизму и приводит к пара-хинонам с выходами, близкими к количественному.
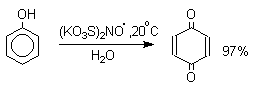
Ароматические амины также гладко окисляются солью Фреми до пара-хинонов.
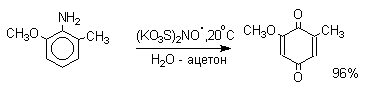
Самый простой и удобный способ получения орто- и пара-бензохинонов состоит в окислении соответственно пирокатехина и гидрохинона. В качестве окислителей можно использовать самые разнообразные реагенты: дихромат натрия, ферроцианид калия, перкислоты, оксид серебра, тетраокись азота и ряд других окислителей.
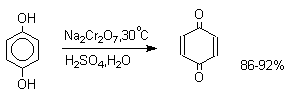
Аналогично ведут себя и пара-аминофенолы.
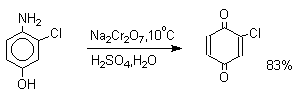
Удовлетворительные результаты при получении орто-бензохинона из пирокатехина достигаются в том случае, если в качестве окислителя используется оксид серебра в эфире в присутствии сульфата натрия для связывания выделяющейся при окислении воды.
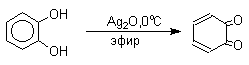
Хиноны, содержащие электроноакцепторные заместители - 2,3,5,6-тетрахлор-1,4-бензохинон ( хлоранил ) или 2,3,5,6-тетрациано-1,4-бензохинон- служат превосходными окислителями для превращения двух- и трехатомных фенолов в хиноны, например:
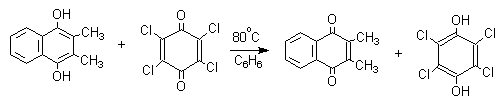
Окисление двухатомных фенолов до 1,4- или 1,2-хинонов осуществляется в несколько стадий, включающих образование анион-радикалов и радикалов. Механизм этой важной в теоретическом отношении модельной реакции будет рассмотрен в разделе "хиноны".
Механизм окисления фенолов, содержащих в обоих орто-положениях и пара-положении алкильные, арильные или алкоксильные группы, тщательно изучен. Е.Мюллер (1953 год) и независимо от него К.Кук обнаружили, что при окислении 2,4,6-три-трет-бутилфенола гексацианоферратом(III) калия K3Fe(CN)6 в бинарной системе бензол-вода в инертной атмосфере образуется устойчивый радикал одновалентного кислорода - три-трет-бутилфеноксил, окрашенный в синий цвет.
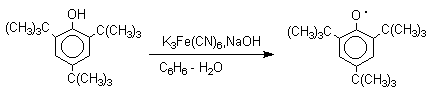
Этот радикал находится в мономерной форме в 0,1 молярном растворе в бензоле или эфире, а также в кристаллическом состоянии. Он очень чувствителен к действию кислорода воздуха, оксида азота (IY), оксида азота (II) и других радикальных частиц. Окисление пространственно затрудненных фенолов до феноксильных (ароксильных) радикалов осуществляется под действием гексацианоферрата (III) калия в бинарной системе бензол-вода, диоксида свинца PbO2, оксида серебра, соли Фреми или другого одноэлектронного окислителя в индифферентной среде, а также электрохимически.
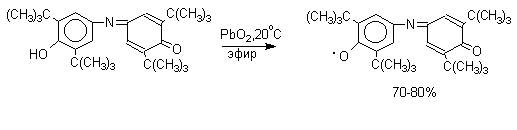
Феноксильные радикалы, содержащие в обоих орто- и пара-положении третичные алкильные группы, мономерны и не проявляют тенденции к образованию димеров в растворе. С другой стороны феноксильные радикалы, содержащие фенильную или алкоксильную группу в орто- или пара-положениях к радикальному центру, проявляют ярко выраженную тенденцию к образованию димеров. Так, например, при окислении 2,4,6-трифенилфенола гексацианоферратом (III) калия получается с выходом 95% димер, который в 0,01 бензольном растворе при 20оС диссоциирован только на 10%. Радикалы с алкоксильной группой в пара-положении более стабильны и 2,6-ди-тре-бутил-4-метоксифеноксил в 3%-ном растворе в бензоле мономерен на 70-75%.
При окислении фенолов образуется несколько различных форм димеров в результате образования новых связей С-С между орто-орто, орто-пара- и пара-пара-положениями исходных радикалов, а также новых С-О связей между атомом кислорода одного радикала и орто-положением другой радикальной частицы. Всего, таким образом, образуется потенциально не менее пяти различных типов димеров, которые находится в равновесии с исходным феноксильным радикалом. Например: для монозамещенного фенола:
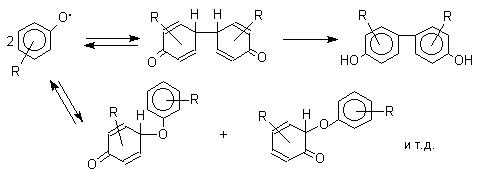
Димеры, возникающие в результате рекомбинации радикалов с образованием новой С-С связи, называются хинолидами. Хинолиды далее изомеризуются с образованием изомерных дигидроксибифенилов. Другой тип димеров, содержащих центральную связь С-О, носит название хиноловых эфиров. Образование хиноловых эфиров свойственно для многих пространственно затрудненных феноксильных радикалов, например, для 2,4,6-трифенилфеноксила; 2,6-ди-трет-бутил-4-фенилфеноксила; 2,6-ди-трет-бутил-4-алкоксифеноксилов и т.д. Хиноловые эфиры могут быть получены прямым окислением соответствующих пространственно затрудненных фенолов.
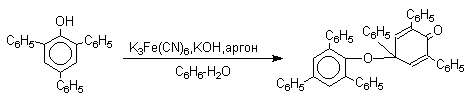
В растворе или при нагревании они диссоциируют на радикалы (см. выше). Хинолидная форма димера образуется в том случае, когда пара-положение или одно из орто-положений феноксила не замещено. Другими словами, хинолиды получаются при окислении 2,4- и 2,6-диалкилфенолов с последующей димеризацией образующихся феноксильных радикалов.
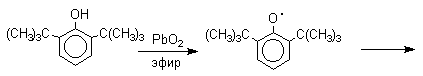
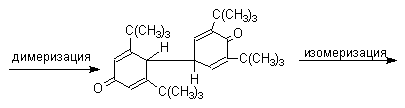
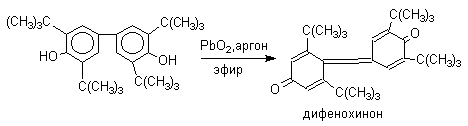
Дальнейшее окисление хинолидного димера, или его таутомерной формы - дигидроксибифенила - приводит к образованию дифенохинона.
Таким образом, производные дифенохинона являются главными конечными продуктами окисления 2,6-диалкилфенолов такими окислителями как: PbO2, Ag2O, K3Fe(CN)6.