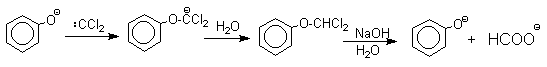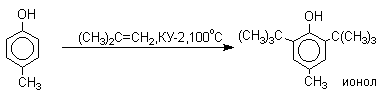
3.5.5. Алкилирование и ацилирование фенолов по Фриделю-Крафтсу
Так как фенолы взаимодействуют с галогенидами алюминия и другими кислотами Льюиса с образованием солей типа ArOAlCl2, прямое их алкилирование в условиях реакции Фриделя-Крафтса провести не удается. Фенолы алкилируют алкенами и спиртами в условиях кислотного катализа. В качестве катализаторов предпочитают использовать серную, фтористоводородную, фосфорную кислоты или катиониты КУ-2, даукс и другие катионообменные смолы. Таким образом, из крезола и изобутилена в промышленности получают пространственно затрудненный фенол - 2,6-ди-трет-бутил-4-метилфенол (ионол), который широко применяется для стабилизации полимеров.
Аналогично из фенола и изопропилового спирта получается 2,4,6-триизопропилфенол.
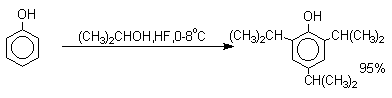
Ацилирование фенолов в классических условиях реакции Фриделя-Крафтса комплексом ацилгалогенида и хлорида алюминия также приводит к неудовлетворительным результатам, так как ацилированию подвергается гидроксильная группа фенола. Более эффективна такая модификация этого метода, когда в качестве ацилирующего агента используется комплекс карбоновой кислоты и трехфтористого бора. Ацильная группа при этом вводится практически исключительно в пара-положение бензольного кольца. Так, например, фенол при взаимодействии с комплексом уксусной кислоты и BF3 дает пара-гидроксиацетофенон с 95%-ным выходом.
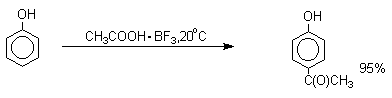
Наиболее общий метод получения гидроксикетонов ароматического ряда основан на перегруппировке Фриса. К.Фрис в 1908 году нашел, что ариловые эфиры карбоновых кислот при нагревании с AlCl3 или AlBr3 перегруппировываются в изомерные орто- или пара-гидроксикетоны. Как правило, в результате перегруппировки образуется смесь орто- и пара-изомеров без примеси мета-изомера.
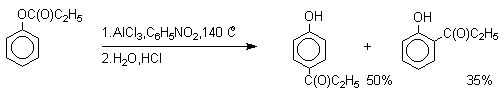
Соотношение орто- и пара-изомеров зависит главным образом от температуры и растворителя. В более жестких условиях преобладает орто-гидроксикетон, а при 20-25оС - пара-гидроксикетон.
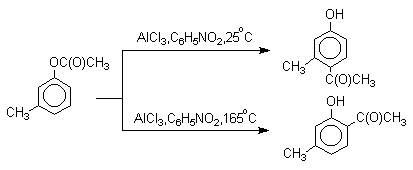
Механизм перегруппировки Фриса, по-видимому, заключается в межмолекулярном ацилировании орто- или пара-положения бензольного кольца арилового эфира комплексом второй молекулы сложного эфира и AlCl3 с образованием ацильного производного гидроксикетона и фенола.
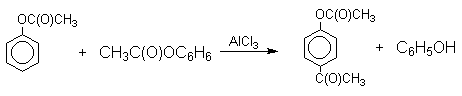
Перегруппировка завершается межмолекулярным переносом ацильной группы к фенолу.
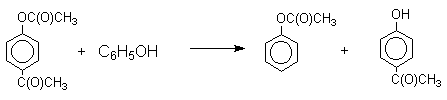
В отличие от самих фенолов их простые эфиры очень легко подвергаются региоселективному ацилированию по Фриделю-Крафтсу в мягких условиях с образованием пара-алкоксиарилкетонов. Наилучшие результаты достигаются при ацилировании простых эфиров фенолов ацилгалогенидами в хлористом метилене при 0оС в присутствии двух молей AlCl3 или AlBr3.
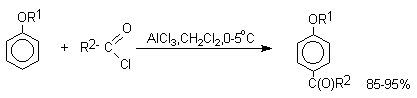
Конденсацию фенолов с фталевым ангидридом в присутствии серной кислоты или хлорида цинка ( А.Байэр, 1874 год ) следует рассматривать как одну из разновидностей реакции ацилирования по Фриделю-Крафтсу. В этом случае две молекулы фенола конденсируются с одной молекулой фталевого ангидрида с образованием производных трифенилметана, называемых фталеинами.
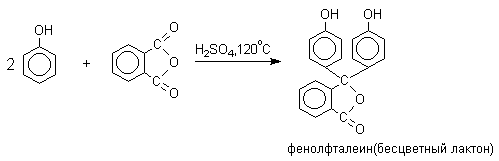
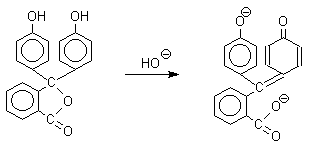
При рН выше 9 водный раствор фенолфталеина окрашивается в малиновый цвет в результате расщепления лактонного цикла и образования дианиона.
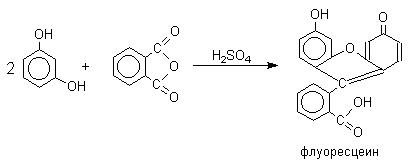
При конденсации фталевого ангидрида с резорцином образуется желто-зеленый флуоресцеин, широко используемый в качестве флуоресцирующего средства.
3.5.6. Формилирование фенолов
В этом разделе из множества методов введения формильной группы в орто- и пара-положения к гидроксильной группе фенолов будут подробно рассмотрены только наиболее синтетически важные реакции: реакции Гаттермана, Вильсмейера-Хаака и Реймера-Тимана.
3.5.6а. Реакция Гаттермана
Попытки введения формильной группы в ароматическое кольцо фенолов, нафтолов и их простых эфиров с помощью СО и HCl ( катализаторы AlCl3 или AlCl3 - Cu2Cl2 ; реакция Гаттермана-Коха ) оказались безуспешными. Поэтому сам Гаттерман предложил метод введения альдегидной группы, в котором в качестве формилирующего агента использовалась смесь безводного HCN и газообразного хлористого водорода (катализатор ZnCl2 , реакция Гаттермана). Формильная группировка вступает в пара-положение к ОН- и RO-группе фенолов или их простых эфиров. Выходы целевых продуктов около 80%.
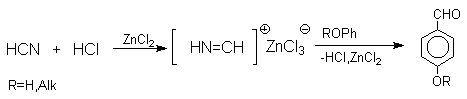
Следует отметить, что истинная природа электрофильной частицы, принимающей участие при введении формильной группы с помощью HCN, HCl и кислоты Льюиса точно не установлена. Для того, чтобы избежать применения ядовитой синильной кислоты, Р.Адамс модифицировал условия реакции, заменив ее цианидом цинка. Это позволило из цианида цинка и HCl получать непосредственно в реакционной смеси HCN и безводный хлористый цинк, играющий роль слабой кислоты Льюиса. Этот метод дает хорошие результаты при формилировании фенолов и их простых эфиров.
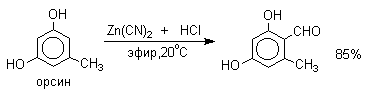
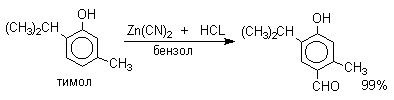
Реакция Гаттермана стала очень популярной после того, как было установлено, что вместо цианидов для введения формильной группы можно использовать нетоксичный и легко доступный симметричный 1,3,5-триазин. Этот метод обеспечивает высокие выходы альдегидов при формилировании алкилбензолов, фенолов, эфиров фенолов, конденсированных углеводородов и гетероциклических соединений.
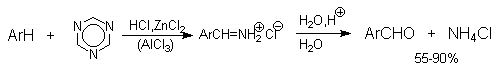
Определенное сходство с реакцией Гаттермана имеет реакция Геша. К.Геш (1915 г) нашел, что реакционноспособные фенолы (обычно двухатомные фенолы, производные резорцина) могут быть успешно ацилированы при взаимодействии с нитрилами и сухим хлористым водородом в присутствии хлорида цинка, как слабой кислоты Льюиса.
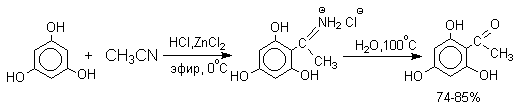
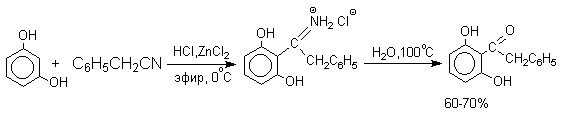
Сам фенол и многие другие одноатомные фенолы в условиях реакции Геша дают не кетоны, а соли иминоэфиров ArOC(R) = N+ H2Cl- .
3.5.6б. Реакция Вильсмейера-Хаака
Для введения альдегидной группы в
ароматическое кольцо, активированное
диалкиламино-, гидрокси- или алкоксигруппой,
эффективна реакция, описанная в 1927 году
А.Вильсмейером и А.Хааком. N-Алкиламиды
муравьиной кислоты -диметилформамид и
N-метилформамид - в присутствии хлорокиси фосфора
являются превосходными региоселективными
формилирующими агентами. С помощью этих
реагентов альдегидная группа вводится в
пара-положение по отношению к имеющейся NR2;
ОН или OR-группе. Эту реакцию можно также
рассматривать как ацилирование, где роль
катализатора - (кислоты Льюиса) выполняют
хлорокись фосфора (POCl3 ), тионилхлорид (SOCl2),
фосген (COCl2). Наиболее эффективна система
ДМФА-POCl3, в которой ДМФА служит и реагентом,
и растворителем. Электрофильным агентом в
реакции Вильсмейера-Хаака является иминиевая
соль, которая образуется при взаимодействии ДМФА
и хлорокиси фосфора, тионилхлорида или фосгена. 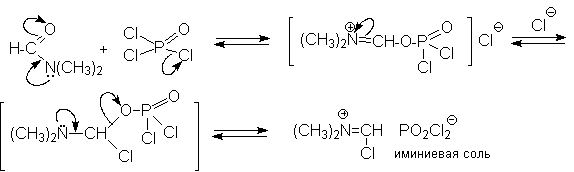
Иминиевая соль при необходимости может быть выделена в индивидуальном виде, например, при взаимодействии ДМФА с фосгеном, где после отщепления СО2 образуется соль (CH3)2N+ =CHCl Cl- . Однако обычно ее не выделяют и используют непосредственно после ее образования.
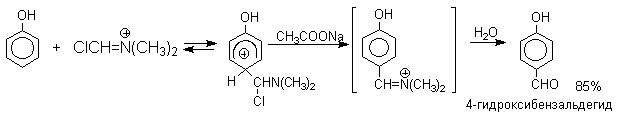
Реакция Вильсмейера-Хаака чрезвычайно проста в экспериментальном отношении и обеспечивает очень высокие выходы ароматических альдегидов, содержащих NR2,OR и ОН группу. Она оказывается практически ценной при формилировании конденсированных ароматических углеводородов - антрацена, азулена, пирена и др., а также разнообразных гетероциклических соединений ряда фурана, тиофена, пиррола, индола. В таблице 2 приведены сводные данные по формилированию фенолов, нафтолов, их простых эфиров.
Таблица 2
| Исходное соединение | Реагент | Катализатор | Условия реакции | Продукт и выход % |
| фенол | ДМФА | POCl3 | ДМФА, 20оС |
4-оксибензальдегид, 85% |
| резорцин | HCN, HCl | ZnCl2 | эфир, -5оС |
2,4-дигидроксибензальдегид, 70% |
| 2-нафтол | CHCl2OCH3 | SnCl4 | CH2Cl2, 20оС |
2-гидрокси-1-формилнафталин, 82% |
| 2-метоксинафталин | ДМФА | POCl3 | ДМФА, 20оС |
2-метокси-1-нафтальдегид, 90% |
| анизол | Cl2CHOCH3 | SnCl4 | CH2Cl2, 0-20оС |
анисовый альдегид,67% |
| анизол | 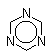 |
AlCl3 | 20оС | анисовый альдегид, 67% |
| анизол | ДМФА | POCl3 | ДМФА | анисовый альдегид, 21% |
| анизол | HCl | Zn(CN)2 | бензол, 40оС |
анисовый альдегид, 94% |
3.5.6в. Реакция Реймера-Тимана
Эта реакция по своему механизму резко отличается от реакций электрофильного замещения в ароматическом кольце фенолов, поэтому ее целесообразно рассматривать отдельно от других способов формилирования фенолов.
Формилирование фенолов по Реймеру-Тиману достигается при нагревании смеси фенола и большого избытка хлороформа с водным раствором гидроксида натрия при 50-70оС. Выходы альдегидов обычно невелики и редко превышают 30%, однако метод исключительно прост и доступен в практическом отношении. Главное достоинство реакции Реймера-Тимана заключается в преимущественном образовании орто-, а не пара-изомеров, как это имеет место для реакций Гаттермана и Вильсмейера-Хаака.
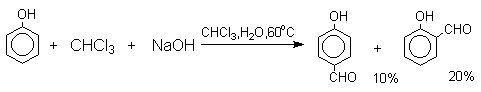
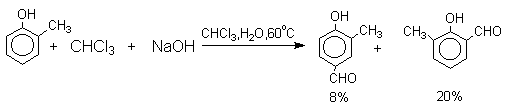
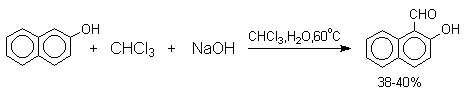
Механизм включает образование дихлоркарбена как интермедиата. Дихлоркарбен : CCl2 выполняет роль электрофильного агента по отношению к феноксид-иону, образующемуся в щелочной среде. Предполагаемый механизм реакции Реймера-Тимана может быть представлен следующей последовательностью превращений:
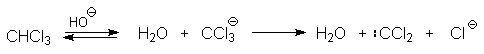
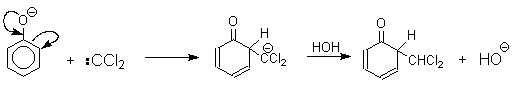
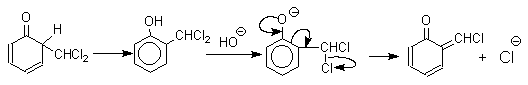
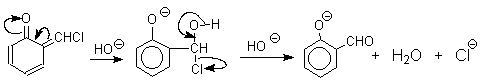
При проведении реакции в D2O более 97% дейтерия включается в формильную группу салицилового альдегида. Это означает, что превращение
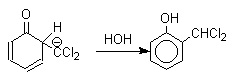
не осуществляется как внутримолекулярный сдвиг.
Хотя анион дихлорметилциклогексадиенона не был выделен в качестве промежуточного продукта реакции Реймера-Тимана, для самого фенола приведенный выше механизм имеет экспериментальные доказательства. Для фенолят-ионов, у которых орто- или пара-положение занято алкильной группой, помимо гидроксибензальдегида другим продуктом реакции всегда оказывается циклогексадиенон, содержащий дихлорметильную группу. Так, например, из пара-крезола получается 2-гидрокси-5-метилбензальдегид и 4-метил-4-дихлорметилциклогексадиен-2,5-он примерно в равных количествах.
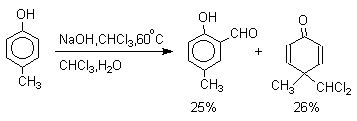
2,4,6-Триметилфенол образует оба возможных изомерных дихлорметилциклогексадиенона, в которых CHCl2 - группа не подвергается гидролизу.
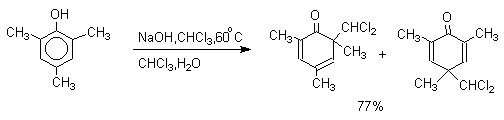
2,6-Ди-трет-бутил-4-метилфенол в этих условиях дает, как и следовало ожидать, только один из двух возможных изомеров.
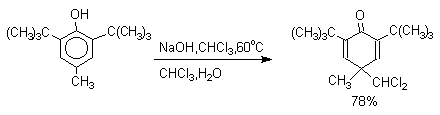
Образование аномальных продуктов служит убедительным подтверждением механизма, предлагаемого для нормального направления реакции, приводящего к образованию альдегида после изомеризации и гидролиза дихлорметильной группы. Реакция протекает только в сильно щелочной среде при наличии фенольного гидроксила, тогда как простые эфиры фенолов и диалкиланилины не формилируются в этих условиях. Выходы альдегидов в реакции Реймера-Тимана невелики, так как большая часть дихлоркарбена гидролизуется в водной щелочи с образованием CO и NaCl. Кроме того, дихлорметиловый эфир фенола, образующийся в результате атаки дихлоркарбена по кислородному атому амбидентного фенолят-иона, нацело гидролизуется до исходного фенола.