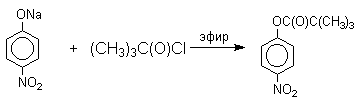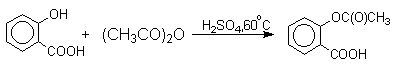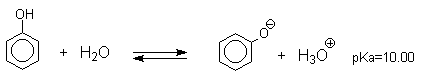
3. Свойства фенолов
3.1. Кислотные свойства фенолов
Несмотря на то, что фенолы по строению подобны спиртам, они являются намного более сильными кислотами, чем спирты. Для сравнения приведем величины рКа в воде при 25оС для фенола (10,00), для циклогексанола (18,00). Из этих данных следует, что фенолы на восемь и более порядков по кислотности превосходят спирты.
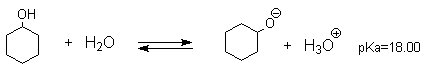
Диссоциация спиртов и фенолов представляет
собой обратимый процесс, для которого положение
равновесия количественно характеризуется
величиной разности свободных энергий 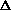 Gо продуктов и исходных
веществ. Для определения влияния строения
субстрата на положение кислотно-основного
равновесия необходимо оценить разницу энергий
между кислотой ROH и сопряженным основанием RO- .
Если структурные факторы стабилизируют
сопряженное основание RO- в большей степени, чем
кислоту ROH, константа диссоциации возрастает и
рКа, соответственно уменьшается. Напротив, если
структурные факторы стабилизируют кислоту в
большей степени, чем сопряженное основание,
кислотность уменьшается, т.е. рКа возрастает.
Фенол и циклогексанол содержат шестичленное
кольцо и поэтому структурно похожи, но фенол в 108
раз более сильная ОН-кислота по сравнению с
циклогексанолом. Это различие объясняется
большим +М эффектом О- в феноксид-ионе. В
алкоголят-ионе циклогексанола отрицательный
заряд локализован только на атоме кислорода и
это предопределяет меньшую стабильность
алкоголят-иона по сравнению с феноксид-ионом.
Феноксид-ион относится к типичным амбидентным
ионам, т.к. его отрицательный заряд делокализован
между кислородом и атомами углерода в орто- и
пара- положениях бензольного кольца. Поэтому для
феноксид-ионов, как амбидентных нуклеофилов,
должны быть характерны реакции не только с
участием атома кислорода, но и с участием атома
углерода в орто- и пара-положениях в бензольном
кольце. Влияние заместителя в бензольном кольце
на кислотность фенолов согласуется с
представлениями об их электронных эффектах.
Электронодонорные заместители понижают, а
электроноакцепторные - усиливают кислотные
свойства фенолов. В таблицах 1 и 1а приведены
данные по кислотности некоторых фенолов в воде
при 25оС.
Gо продуктов и исходных
веществ. Для определения влияния строения
субстрата на положение кислотно-основного
равновесия необходимо оценить разницу энергий
между кислотой ROH и сопряженным основанием RO- .
Если структурные факторы стабилизируют
сопряженное основание RO- в большей степени, чем
кислоту ROH, константа диссоциации возрастает и
рКа, соответственно уменьшается. Напротив, если
структурные факторы стабилизируют кислоту в
большей степени, чем сопряженное основание,
кислотность уменьшается, т.е. рКа возрастает.
Фенол и циклогексанол содержат шестичленное
кольцо и поэтому структурно похожи, но фенол в 108
раз более сильная ОН-кислота по сравнению с
циклогексанолом. Это различие объясняется
большим +М эффектом О- в феноксид-ионе. В
алкоголят-ионе циклогексанола отрицательный
заряд локализован только на атоме кислорода и
это предопределяет меньшую стабильность
алкоголят-иона по сравнению с феноксид-ионом.
Феноксид-ион относится к типичным амбидентным
ионам, т.к. его отрицательный заряд делокализован
между кислородом и атомами углерода в орто- и
пара- положениях бензольного кольца. Поэтому для
феноксид-ионов, как амбидентных нуклеофилов,
должны быть характерны реакции не только с
участием атома кислорода, но и с участием атома
углерода в орто- и пара-положениях в бензольном
кольце. Влияние заместителя в бензольном кольце
на кислотность фенолов согласуется с
представлениями об их электронных эффектах.
Электронодонорные заместители понижают, а
электроноакцепторные - усиливают кислотные
свойства фенолов. В таблицах 1 и 1а приведены
данные по кислотности некоторых фенолов в воде
при 25оС.
Таблица 1.
Величины рКа орто-, мета- и пара-замещенных фенолов в воде при 25оС
| Заместитель | орто | мета | пара |
| H | 10.00 | 10.00 | 10.00 |
| CH3 | 10.29 | 10.09 | 10.26 |
| C(CH3)3 | 10.62 | 10.12 | 10.23 |
| C6H5 | 10.01 | 9.64 | 9.55 |
| OCH3 | 9.98 | 9.65 | 10.21 |
| COOC2H5 | 9.92 | 9.10 | 8.34 |
| F | 8.73 | 9.29 | 9.89 |
| Cl | 8.56 | 9.12 | 9.41 |
| Br | 8.45 | 9.03 | 9.37 |
| I | 8.51 | 9.03 | 9.33 |
| HCO | 8.37 | 8.98 | 7.61 |
| CN | 6.86 | 8.61 | 7.97 |
| NO2 | 7.23 | 8.36 | 7.15 |
Таблица 1а
Величины рКа некоторых полизамещенных фенолов и нафтолов
| пентахлорфенол | 5.26 |
| пентафторфенол | 5.5 |
| 1-нафтол | 9.39 |
| 2-нафтол | 9.63 |
| 2,4-динитрофенол | 4.07 |
| 2,4,6-тринитрофенол | 0.42 |
3.2. Таутомерия фенолов
Естественно, что между амбидентными феноксид и енолят-ионами существует определенная аналогия. Фенол также является аналогом енола и между ним и его кето-формами (2,4- и 2,5-циклогексадиенами) должны существовать отношения, подобные тем, которые наблюдаются для равновесия кето- и енольной форм кетонов.
Для самого фенола энтальпия этого равновесия
составляет величину порядка +23 Ккал/моль, что
соответствует логарифму константы равновесия в
уравнении Гиббса порядка 9,5  2,5, т.е.
равновесие целиком смещено в сторону фенольной
формы. Таким образом, соотношение двух
таутомерных форм здесь полностью обратно тому,
которое наблюдается для кетонов, где преобладает
кето-форма. Это различие не удивительно, если
принять во внимание ароматическую стабилизацию
бензольного кольца фенола. Для других одно- и
двухатомных фенолов бензольного ряда также не
удается наблюдать прототропную таутомерию. Для
полициклических конденсированных ароматических
соединений различие в термодинамической
устойчивости фенольной и диеноновой форм резко
уменьшается, и для 1-нафтола фенольная форма на 12,5
Ккал/моль стабильнее диенона, тогда как для
9-гидроксиантрацена кетоформа антрона уже
оказывается термодинамически более стабильной
формой.
2,5, т.е.
равновесие целиком смещено в сторону фенольной
формы. Таким образом, соотношение двух
таутомерных форм здесь полностью обратно тому,
которое наблюдается для кетонов, где преобладает
кето-форма. Это различие не удивительно, если
принять во внимание ароматическую стабилизацию
бензольного кольца фенола. Для других одно- и
двухатомных фенолов бензольного ряда также не
удается наблюдать прототропную таутомерию. Для
полициклических конденсированных ароматических
соединений различие в термодинамической
устойчивости фенольной и диеноновой форм резко
уменьшается, и для 1-нафтола фенольная форма на 12,5
Ккал/моль стабильнее диенона, тогда как для
9-гидроксиантрацена кетоформа антрона уже
оказывается термодинамически более стабильной
формой.
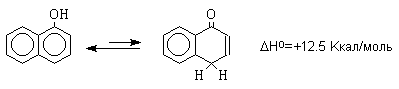
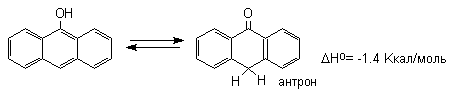
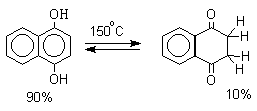
Для 9-гидроксиантрацена известны обе формы, причем в неполярных растворителях (петролейный эфир, бензол, ССl4) равновесие сдвинуто в сторону антрона (К20=400 в бензоле). Устойчивость таутомерных кето-форм возрастает при переходе к полиатомным фенолам. Так, при плавлении 1,4-дигидроксинафталина получается равновесная смесь, содержащая 10%-дикетоформы.
Длительное время не предпринималось попыток фиксации или стабилизации кето-формы фенолов ряда бензола и нафталина и проблема таутомерии фенолов не привлекала внимания исследователей. В 1968 году В.А.Коптюг с сотрудниками предложил простой и чрезвычайно эффективный способ стабилизации кето-формы разнообразных фенолов с помощью сильных кислот Льюиса- хлорида или бромида алюминия. Эти жесткие кислоты Льюиса связывают жесткий карбонильный кислород кето-формы в очень стабильный комплекс, который может быть зафиксирован с помощью ПМР- и ИК-спектроскопии. При этом для 1-нафтола, 2-нафтола и фенолов бензольного ряда, содержащих хотя бы одну алкильную группу в мета-положении к гидроксилу, равновесие в неполярной среде нацело смещается в сторону кето-формы.
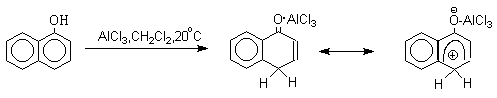
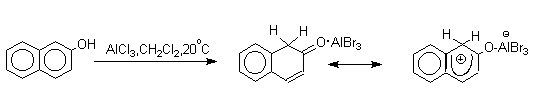
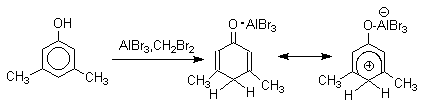
Эти комплексы можно рассматривать также как биполярные ионы, т.е. как гидроксиаренониевые ионы, образующиеся при протонировании фенолов по бензольному кольцу. Для них должны быть характерны реакции, типичные для аренониевых ионов. Действительно, комплекс 2-нафтола с хлоридом алюминия может быть использован в качестве электрофильного агента в электрофильном ароматическом замещении в бензольном кольце.
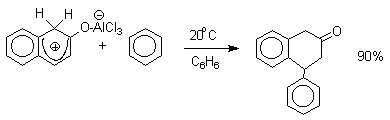
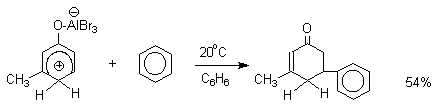
Аналогично реагирует и комплекс мета-крезола с AlBr3.
Кето-енольная таутомерия лежит в основе
замещения фенольного гидроксила на аминогруппу,
которое происходит при нагревании 1- или
2-гидроксинафталина, сульфопроизводных  - и
- и  -нафтолов, 6- или
8-гидроксихинолинов и других
гидроксипроизводных нафталина, антрацена,
хинолина с водным раствором сульфита или
гидросульфита аммония при 130-150оС.
-нафтолов, 6- или
8-гидроксихинолинов и других
гидроксипроизводных нафталина, антрацена,
хинолина с водным раствором сульфита или
гидросульфита аммония при 130-150оС.
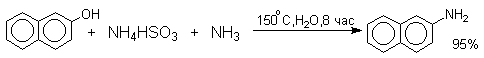
Эта реакция, открытая Г.Бухерером в 1904 году, применима к одноатомным фенолам ряда нафталина, антрацена и других конденсированных ароматических систем, но в нее практически не вступают одноатомные фенолы бензольного ряда.
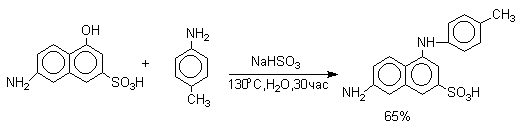
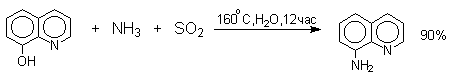
В ряду производных бензола в реакцию вступают многоатомные фенолы, содержащие две или три гидроксильные группы в мета-положении к друг другу:
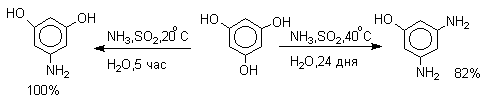
Для реакции Бухерера предложен, но строго не
доказан механизм, в котором сульфит- или
гидросульфит-ион присоединяется в -положение кето-формы нафтола с
образованием -кетосульфокислоты
в качестве интермедиата, который в отдельных
случаях был выделен в индивидуальном виде.
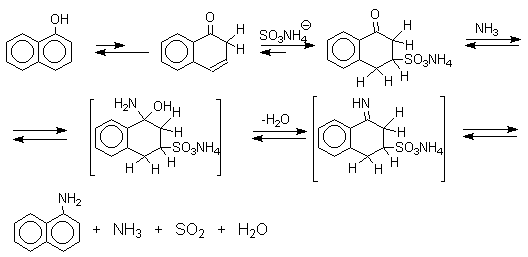
Последующее аминирование и отщепление сернистой кислоты приводит к амину. Реакция обратима и при обработке 1- или 2-амино-нафталина и их производных гидросульфитом натрия в воде при 130-150оС они превращаются в соответствующие фенолы, например:
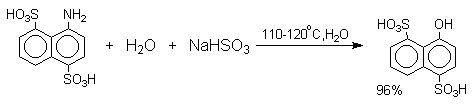
3.3. С- и О-Алкилирование амбидентных фенолят-ионов
Феноксид-ионы, как типичные амбидентные анионы, способны к реакциям О- и С-алкилирования. В большинстве случаев феноксид-ионы подвергаются региоселективному О-алкилированию независимо от природы радикала и уходящей группы алкилирующего агента. Самые разнообразные жесткие и мягкие алкилирующие агенты дают эфиры фенолов в реакциях со щелочными фенолятами. Наилучшие результаты для О-алкилирования достигаются в диполярных апротонных растворителях, хорошо сольватирующих катионы щелочных металлов.
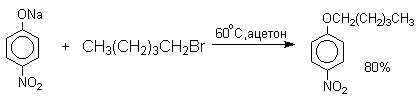
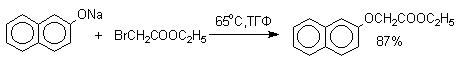
Если же растворитель избирательно сольватирует кислородный центр феноксид-иона, открывается возможность для С-алкилирования. Кислородный центр феноксид-ионов подвергается избирательной сольватации с помощью водородной связи в воде, трифторэтаноле, феноле. В этих растворителях удается осуществить С-алкилирование феноксид-иона под действием таких мягких алкилирующих агентов, как аллилгалогениды и бензилгалогениды.
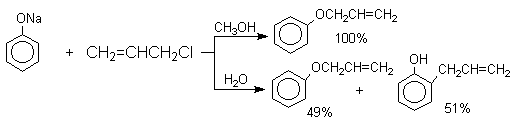
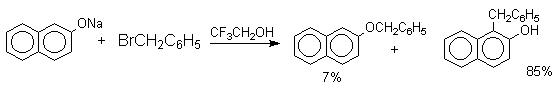
Кислородный центр аниона блокируется в результате избирательной сольватации и алкилирующий агент атакует другой нуклеофильный центр амбидентного аниона - атом углерода в орто-положении по отношению к атому кислорода. Аналогичный результат может быть достигнут за счет блокирования кислородного центра противоионом в сложном ассоциате ионных пар щелочных фенолятов и нафтолятов. Действительно, при алкилированиии фенолята натрия в бензоле аллилбромидом образуется 2-аллилфенол, тогда как в ацетоне в тех же условиях получается только аллиловый эфир фенола.
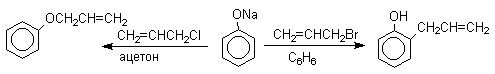
По той же причине алюминиевые соли фенолов подвергаются С-алкилированию в орто-положение бензольного кольца.
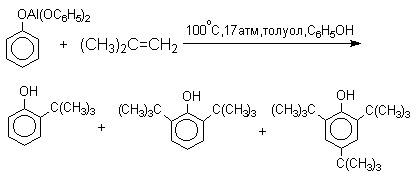
Эта реакция имеет практическое значение для синтеза стерически затрудненных фенолов, используемых в качестве стабилизаторов полимеров против термоокислительной деструкции. Фенолы, подобно карбоновым кислотам, подвергаются селективному О-метилированию при действии диазометана в эфирном растворе. Напомним, что спирты реагируют с диазометаном только в присутстсвии катализаторов - кислот Льюиса.
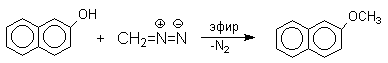
3.4. Этерификация фенолов
Ариловые эфиры карбоновых кислот нельзя получать прямой этерификацией фенолов карбоновыми кислотами. Обратимая реакция фенола с уксусной кислотой эндотермична в отличие от реакции этерификации спиртов, которая экзотермична.
![]()
![]()
Ариловые эфиры карбоновых кислот получают ацилированием фенолов или их Na-, K-солей галогенангидридами или ангидридами кислот.