1.Йодбензол. ЛАБ с. 200
Анилин 9,3 г
HCl конц. 25 мл
NaNO2 8 г
KI 20 г
По этой методике можно получать изомерные
йодтолуолы, йоданизолы и йоддифениловые эфиры.
2.о-Йодбензойная кислота.
Прямое диазотирование.
Антраниловая кислота 6,3 г
H2SO4 50% 60мл
NaNO2 3,4 г в 6 мл воды
В стакан емкостью 100 мл, снабженный
механической мешалкой и капельной воронкой,
вносят 6,3 г антраниловой кислоты и 60 мл 50% H2SO4.
Растворяют при нагревании, а затем при
перемешивании охлаждают до 0оС. При
перемешивании и внешнем охлаждении из капельной
воронки медленно добавляют раствор 3,4 г NaNO2
в 6 мл воды. Добавление ведут с такой скоростью,
чтобы температура реакционной смеси не
поднималась выше 5оС и не выделялись окислы
азота. Избыток HNO2 удаляют добавлением
мочевины (до отрицательной пробы по
йодкрахмальной бумажке). Профильтрованный при
наружном охлаждении диазораствор прибавляют к
холодному раствору 9 г KI в 50 мл 3% -ной H2SO4,
разбавляют вдвое холодной водой и оставляют на
ночь. На следующий день реакционную смесь при
перемешивании нагревают на кипящей водяной бане
с обратным холодильником до отрицательной пробы
с 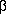 -нафтолом. Нагревание
проводят примерно в течение 2-х часов. По
охлаждении выпавшую о-йодбензойную кислоту
отфильтровывают, полученный осадок растворяют в
минимальном количестве раствора Na2SO3,
полученный раствор профильтровывают и выливают
при перемешивании в большой объем разбавленной
(10%-ной) HCl. Выпавший осадок отфильтровывают, сушат
на воздухе. Выход о-йодбензойной кислоты 10,2 г
(89% ), т. пл. 160-161оС.
-нафтолом. Нагревание
проводят примерно в течение 2-х часов. По
охлаждении выпавшую о-йодбензойную кислоту
отфильтровывают, полученный осадок растворяют в
минимальном количестве раствора Na2SO3,
полученный раствор профильтровывают и выливают
при перемешивании в большой объем разбавленной
(10%-ной) HCl. Выпавший осадок отфильтровывают, сушат
на воздухе. Выход о-йодбензойной кислоты 10,2 г
(89% ), т. пл. 160-161оС.
3.п-Бромтолуол.СОП ч. 3 с. 137
а)Бромистая медь. СОП ч.3 с. 468 ; ПОХ с. 169
Медный купорос 30 г
Калия бромид 20,2 г
Натрий сернистокислый 7,6 г
30 г Медного купороса и 20,2 г бромида калия
растворяют в 100 мл теплой воды, к этому раствору
при перемешивании прибавляют порциями 7,6 г
сернистокислого натрия ( до обесцвечивания
раствора ), затем смесь охлаждают. Бесцветный
тяжелый осадок бромистой меди отфильтровывают
на воронке Бюхнера, промывают водой, тщательно
отжимают и высушивают на воздухе в течение ночи.
Выход 16 г ( 90% ).
В стакане емкостью 250 мл к 100 мл воды прибавляют
при перемешивании 10,75 мл конц. H2SO4, а
затем, не охлаждая, 10,75 г п-толуидина и
нагревают смесь, если нужно, до полного
растворения амина. Полученный раствор быстро
охлаждают в ледяной бане при интенсивном
перемешивании ( чтобы выпали мелкие кристаллы
соли амина ) до 5-10оС, диазотируют раствором
7 г NaNO2 в 12,5 мл воды, поддерживая температуру
реакционной смеси в интервале 10-15оС, и
оставляют при охлаждении на 15-20 мин. Затем
полученный диазораствор обрабатывают сухой
мочевиной (осторожно, вскипание!) до
отрицательной пробы по йодкрахмальной бумажке, и
вносят его порциями по 10-15 мл при перемешивании в
раствор 8 г бромистой меди ( Cu2Br2), 2,5 г
KBr и 0,1 г порошкообразной меди ( приготовление см.
ЛАБ с. 186) в 10 - 15 мл 48%-ной HBr и оставляют на ночь. На
другой день, если проба на диазоний с -нафтолом положительная, реакционную
смесь нагревают с обратным холодильником до
отрицательной пробы с -нафтолом,
подщелачивают раствором NaOH и отгоняют
п-бромтолуол с паром. п-Бромтолуол отделяют,
водный слой экстрагируют бензолом и вытяжки
объединяют с основной порцией вещества.
Бензольный раствор п-бромтолуола промывают
несколько раз конц. H2SO4 (пока она не
перестанет темнеть ), затем водой и высушивают
над CaCl2. Бензол отгоняют на роторном
испарителе, а остаток перегоняют в вакууме.
Выход 12 – 12,5 г ( 70% ), т. кип. 183-185о/755 мм рт.
ст., т. пл. 25-26оС.
4.о-Бромхлорбензол.СОП ч.3 с. 467
о-Хлоранилин 12,75 г
HBr 48%-ная 38 мл
Нитрит натрия 7 г
Медь бромистая (CuBr) 7,9 г
В колбу на 250 мл помещают смесь 12,75 г
о-хлоранилина и 30 мл 48%-ной HBr. Колбу устанавливают
в бане со льдом и, прибавляя к содержимому колбы
лед, охлаждают его до 0оС. Затем к смеси
быстро при перемешивании приливают раствор 7 г
нитрита натрия в 12,5 мл воды, следя за тем, чтобы
температура реакционной смеси не превышала 10оС,
и при необходимости добавляя кусочки льда.
Последние 2-3 мл раствора нитрита натрия в воде
прибавляют с осторожностью и до тех пор, пока в
растворе не будет обнаружен избыток HNO2
(контроль по йодкрахмальной бумажке).
Одновременно смесь 7,9 г CuBr и 8 мл 48%-ной HBr
нагревают до кипения в круглодонной 3-х горлой
колбе на 500 мл, снабженной обращенным вниз
холодильником, к которому в качестве приемника
присоединена колба на 200 мл, трубкой для ввода
пара, которая перекрывается винтовым зажимом и
делительной воронкой. Примерно ? часть раствора
диазония переносят не фильтруя в делительную
воронку и немедленно приливают к раствору CuBr в HBr,
который нагревают до кипения на пламени горелки.
Приливание ведут с такой скоростью, чтобы
кипение было непрерывным. Следующую порцию
охлажденной соли диазония добавляют в капельную
воронку, не прерывая прибавления его к CuBr. Весь
раствор добавляют за 15-20 мин, в течение этого
времени большая часть вещества перегоняется с
водяным паром. Когда прибавление закончено, кран
на воронке перекрывают, открывают зажим на
паропроводе и в смесь пропускают сильную струю
пара (Очки!) до тех пор, пока не перестанет
перегоняться органическое вещество. Всего
собирают примерно 100 – 150 мл дистиллята.
Органический слой отделяют от дистиллята и
промывают конц. H2SO4 порциями по 2 – 5
мл до тех пор, пока кислота не начнет при этом
лишь слабо окрашиваться ( обычно промывают 3 – 4
раза). Затем полученную маслянистую жидкость
промывают 1 раз водой ( 10 мл), 2 раза 5%-ным раствором
NaOH ( 2 х 5 мл) и наконец еще 1 раз водой (10 мл).
Высушивают над прокаленным CaCl2 и
перегоняют. Выход о-бромхлорбензола 17 – 18,5 г
(89 – 95% от теорет.), т. кип. 199 – 201 оС / 742 мм.
Примечания : а) Дистиллят, полученный при
перегонке с паром, подщелачивают и перегоняют с
паром еще раз. о-Бромхлорбензол отделяют от воды
и разбавляют в 2-3 раза бензолом. Дальнейшая
очистка по методике. б) По этой методике можно
получить м-хлорбромбензол, м-дибромбензол с
выходом 80-87%, а также о-броманизол. Последний
нельзя промывать конц. H2SO4. в)
Приготовление бромистой меди приведено в
синтезе п-бромтолуола.
5.п-Нитройодбензол. Лабораторные
методики вып. 3 с. 49
п-Нитроанилин 1 г
H2SO4 конц. 1,5 г (0,9 мл)
NaNO2 0,5 г
KI 2 г
Г Е Т Е Р О Ц И К Л Ы
1. Хинолин. ЛАБ с. 222
Синтез рекомендуется проводить в 3-х горлой колбе
с мешалкой.
Синтез проводится в вытяжном шкафу.
Нитробензол 10 г
Анилин 15,5 г
Глицерин 50 г
H2SO4 конц. 24 мл
Добавить в реакционную смесь FeSO4 на кончике
шпателя.
NaOH (40%-ный раствор)
NaOH твердый
Эфир 90 мл
HCl конц. 25 мл
ZnCl2 15 г
2. 2,3 - Диметилиндол.
Фенилгидразин солянокислый 7,5 г
Метилэтилкетон 4,5г
CH3COOH лед. 25 мл
В 3-х горлую колбу с обратным холодильником и
термометром помещают 4,5 г метилэтилкетона, 25 мл
ледяной CH3COOH и 7,5 г солянокислого
фенилгидразина и нагревают до 90оС. Через
15-20 мин смесь саморазогревается и вскипает CH3COOH.
После того как самопроизвольное вскипание
прекращается, нагревают еще 30 мин при 90оС.
Смесь охлаждают, выливают в лед (75 г), осадок
промывают 125 мл воды, растворяют в 75 мл эфира.
Эфирный раствор промывают 20 мл 10% -ной HCl, двумя
порциями по 20 мл 5%-ного NaOH и двумя порциями воды
по 20 мл. Эфирный экстракт высушивают Na2SO4,
эфир отгоняют на роторном испарителе. Получают 6,3
г ( 90% ) 2,3-диметилиндола с т. пл. 101 – 104 оС.
Перекристаллизовывают из петролейного эфира.
Получают 5,7 г ( 80% ) 2,3-диметилиндола с т. пл. 102 – 106 оС.
3.2 - Фенилиндол.
а) Гат. С. 10
Ацетофенон 6 г (5,6 мл)
Фенилгидразин 5,4 г
Фосфорная кислота 20 мл
Фосфорный ангидрид 40 г
Приготовление фенилгидразона ацетофенона.
В круглодонную колбу на 100 мл помещают 6 г
ацетофенона и 5,4 г свежеперегнанного
фенилгидразина. Смесь нагревают на водяной бане
при 100оС 1 час, охлаждают и растирают до тех
пор, пока она не затвердеет. Затвердевшую массу
растирают с 10 мл этанола, продукт
отфильтровывают, промывают на фильтре
охлажденным до 0о этанолом ( 10 мл).
Фенилгидразон ацетофенона имеет т. пл. 105оС.
Выход 9,5 - 10 г.
Приготовление полифосфорной кислоты.
В 3-х горлую колбу с мешалкой и термометром
приливают 20 мл фосфорной кислоты. При
перемешивании добавляют порциями 40 г фосфорного
ангидрида. После внесения всего фосфорного
ангидрида нагревают смесь при перемешивании 30
мин при 80оС.
Получение 2-фенилиндола.
К полученной полифосфорной кислоте прибавляют
фенилгидразон ацетофенона ( ~ 10г ) и перемешивают
смесь 1 час при 100оС. По охлаждении до 80оС
выливают на лед ( 400 г ) и отфильтровывают выпавший
осадок серого цвета, промывают на фильтре водой,
перекристаллизовывают из этанола, применяя
активированный уголь ( расход этанола 4 мл на 1 г
вещества ).
Выход 5,4 г , т. пл. 185 – 186 оС.
б) Органикум, 1965 г, с. 562
В маленькую широкогорлую колбу Эрленмейера к
смеси 0,05 моля ацетофенона и 0,055 моля
фенилгидразина прибавляют 20 г полифосфорной
кислоты *. Перемешивая термометром смесь
медленно нагревают, пока не заметят резкого
повышения температуры, вызванного началом
реакции. Теперь температуру поддерживают в
пределах 175-190оС, охлаждая колбу при
необходимости на кипящей водяной бане. После
окончания реакции дают смеси охладиться и
разбавляют 50 мл холодной воды. Экстрагируют
эфиром раствор, высушивают Na2SO4 и
растворитель отгоняют. Остаток очищают
кристаллизацией из этанола. Выход 76 % , т. пл. 188оС.
*) Полифосфорная кислота. К 8 мл 85% - ной H3РO4
нагретой на плитке в конической колбе, порциями
добавляют 16 г P2O5 при перемешивании до
образования прозрачной, желтоватой
сиропообразной массы. Это количество все
использовать для реакции.
4. 2-Фенилбензимидазол. Гет. С. 82
о-Фенилендиамин (студ.) 10,8 г
Бензойная кислота 12,2 г
Фосфорная кислота 20 мл
Фосфорный ангидрид 30 г
Смешивают 10,8 г о-фенилендиамина и 12,2 г
бензойной кислоты с 50 г полифосфорной кислоты (
получение ПФК дано в предыдущем синтезе
2-фенилиндола). Образовавшуюся пасту медленно
нагревают при перемешивании до 190-200оС и
полученный раствор при этой температуре
выдерживают 2-3 часа. Затем массу охлаждают до 100оС
и выливают тонкой струйкой в 50 мл энергично
перемешиваемой воды. Осадок отфильтровывают,
промывают небольшим количеством воды и
обрабатывают избытком 10%-ного раствора Na2CO3.
Осадок отфильтровывают, промывают водой и сушат
при 100оС. Выход сырого продукта 18,5 г ( 95% ).
После перекристаллизации из спирта получают 15,5 г
продукта с т. пл. 294 - 295,5оС.
5. 2-Метилбензоксазол. ЛАБ с. 218
о-Аминофенол 5 г
Уксусный ангидрид 12,5 г
Примечание : 1) 2-метилбензоксазол имеет nD20
= 1,5500 и т. кип. ~ 85о/20 мм.
6. Бензоксазол. Гет. с. 90
о-Аминофенол 18 г
HCOOH 99,5 % 10 мл
В круглодонной колбе емкостью 50 мл кипятят с
обратным холодильником в течение 1 часа смесь 18 г о-аминофенола
и 10 мл 99,5 % HCOOH. Затем, заменив обратный
холодильник насадкой Вюрца с нисходящим
холодильником, реакционную смесь перегоняют,
собирая фракцию с т. кип. 140-210оС. Дистиллят
встряхивают с насыщенным раствором поташа для
нейтрализации избытка кислоты. Бензоксазол
экстрагируют эфиром (25 мл), вытяжку сушат поташом,
растворитель отгоняют и остаток перегоняют,
собирая фракцию с т. кип. 181-183оС.
Бензоксазол перегоняется в виде почти
бесцветной жидкости (d420=1,17), при
желании его можно перегнать в вакууме. Выход 8,1 г
(41%). При длительном хранении бензоксазол
образует крупные бесцветные призмы с т. пл. 30 - 31оС.
7. Бензотриазол. Гет. с. 87
о-Фенилендиамин 5,4 г
CH3COOH 12 мл
NaNO2 3,5 г
К раствору 5,4 г о-фенилендиамина в смеси 12 мл CH3COOH
и 25 мл воды, помещенному в стакане емкостью 250 мл,
добавляют при перемешивании раствор 3,5 г NaNO2
в 10 мл воды. Происходит самопроизвольное
разогревание реакционной смеси, температура ее
достигает ~ 70оС и окраска становится
оранжево-красной. Раствор охлаждают на ледяной
бане и нейтрализуют добавлением 2н NH4OH до рН
7, продолжая перемешивать смесь до выпадения
кристаллического осадка. Коричневый осадок
отфильтровывают, промывают ледяной водой ( 4 х 25
мл) и сушат при 70-80оС, а затем
перекристаллизовывают из бензола ( на 1 г
вещества расходуется около 5 мл бензола) с
применением активированного угля. Выход 4 – 4,6 г
(67 – 77%). Бесцветные иглы с т. пл. 98 – 99оС.
Бензотриазол можно также очистить перегонкой в
вакууме, собирая фракцию с т. кип. 156–159 о (2
мм рт. ст.).
8.2-Метилбензимидазол.Гет. с. 78
о-Фенилендиамин 10,8 г
CH3COOH 9 мл
Смесь 10,8 г о-фенилендиамина, 9 мл CH3COOH и
1-2 мл конц. HCl, помещенную в круглодонную колбу
емкостью 50 мл, кипятят с обратным холодильником
1,5-2 часа , разбавляют 12 мл воды, добавляют 1 г
активированного угля и кипятят еще 15-20 минут.
Уголь отфильтровывают, а фильтрат осторожно
подщелачивают при энергичном перемешивании и
охлаждении 20 % -ным раствором NH4OH до
появления в реакционной смеси слабого запаха
аммиака. Выпавший осадок отфильтровывают,
тщательно промывают ледяной водой (4 х 5 мл) и
сушат при 100 – 110оС. Выход 10-10,6 г (75-80%), т. пл.
173-174оС. Полученный продукт слегка окрашен,
его можно использовать для дальнейших синтезов
без дополнительной очистки, но при необходимости
2-метилбензимидазол очищают перекристаллизацией
из воды с применением активированного угля.
Получают белоснежные иголочки с т. пл. 176 – 177 оС.
9.4,5-Дифенилимидазол. Гет. с. 74-75
Бензил 5,25 г
Уротропин 0,65 г
CH3COONH4 3г
CH3COOH 125 мл
Раствор 5,25 г бензила, 0,65 г уротропина и 3 г CH3COONH4
в 125 мл CH3COOH кипятят с обратным
холодильником 1 час. По охлаждении реакционную
массу выливают в 1,25 л воды. Полученный раствор
обесцвечивают активированным углем, фильтруют,
фильтрат нейтрализуют водным раствором аммиака.
Выпавший осадок 4,5-дифенилимидазола отделяют
фильтрованием, промывают на фильтре водой ( 4 х 30
мл), высушивают при 100 оС.
Выход 5 г ( 91%), бесцветные призмы с т. пл. 231 – 232оС
(из диоксана).
10. 3,5-Диметилпиразол. Гет. с.
Гидразин сернокислый 13 г
NaOH 10%-ный раствор 80 мл
Ацетилацетон 10 г
В 3-х горлой колбе на 250 мл, снабженной мешалкой,
термометром и капельной воронкой (Не допускать
герметичности прибора!) готовят раствор 13 г
сернокислого гидразина в 80 мл 10%-ного раствора NaOH
и, поместив колбу в баню со льдом, охлаждают
раствор до 15оС. Поддерживая эту
температуру, к раствору в течение 30 мин
прибавляют по каплям 10 г ацетилацетона, после
чего смесь перемешивают при 15оС еще 1 час.
Затем прибавляют 40 мл воды для растворения
осадка неорганических солей и переносят в
делительную воронку, экстрагируя образовавшийся
пиразол один раз 25 мл эфира, а затем еще 4 раза
порциями по 10 мл. Эфирные вытяжки объединяют,
промывают насыщенным раствором NaCl и сушат
безводным поташом. После отгонки эфира получают
3,5-диметилпиразол в виде белых или слегка
желтоватых кристаллов с т. пл. 107-108оС. Выход
7,4 - 7,8 г ( 77-81%).
11. 4(5) – Фенилимидазол. Гет. с.
Синтез фенацилбромида приведен в
разделе"Альдегиды и кетоны ". Лакриматор !
Синтез выполнять под тягой.
Фенацилбромид 8 г
Формамид 50 мл
Смесь 8 г фенацилбромида (Лакриматор ! Тяга!)
и 50 мл формамида кипятят в колбе с обратным
воздушным холодильником 2 часа. При этом сначала
образуется ярко-красный раствор, который через
10-15 минут светлеет и до конца реакции остается
светло-оранжевым. По охлаждении его обрабатывают
200 мл горячей разбавленной HCl ( 1: 1), при
необходимости кипятят с углем, фильтруют и
фильтрат подщелачивают раствором аммиака до рН
7-8. Выпавший 4(5)-фенилимидазол отделяют и очищают
последовательной перекристаллизацией из воды и
бензола, получая бесцветные пластинки с т. пл. 128
– 129оС.
Выход 5,2 г (90%).