Медь и её соединения
Дихлорид меди
CuCl2(г). Термодинамические свойства газообразного
дихлорида меди в интервале температур 100 - 6000 K приведены в табл.CuCl2.
Молекулярные постоянные, принятые для
расчета термодинамических функций CuCl2,
приведены в табл.Cu.10. Cложность
интерпретации спектров молекулы CuCl2
[84VAN/DEV], [77DIE/EMM], [79PAP], а также
необходимость использования высоких уровней abinitio расчетов [68LOH, 71SMI, 78GAR/HIL, 80COR/HEH, 81ITO, 84HA/NGU, 89BAU/ROO], приводили к противоречивым выводам относительно
её строения и системы электронных состояний. Однако, выполненные детальные
исследования электронных состояний и колебательно-вращательных спектров CuCl2 и abinitio расчеты [2001WAN/WAN, 2000BOS/CRO, 92YAN/QIN] позволили снять все противоречия. На основании
результатов этих работ принимается, что молекула CuCl2
в основном электронном состоянии Х2Pg(3/2) имеет линейную конфигурацию (симметрия D¥h) (r(Cu-Cl) = 2.0341(3) Å).
Энергия возбуждения второго компонента спин-орбитально расщепления, А2Pg(1/.2), равна 178 см‑1. Оказалось, что
и в возбужденных электронных состояниях молекула также является линейной. При
этом значения межъядерного расстояния и частот колебательного спектра близки к
соответствующим значениям в основном состоянии. Момент инерции для основного
расстояния рассчитан из значения Be = 0,055106(3) см‑1,
измеренного на основе анализа 1782 колебательно-вращательных уровней авторами
работы [2000BOS/CRO].
Погрешность момента инерции равна 0.0055% его значения. По данным этой же
работы приняты значения частот колебательного спектра для основного состояния.
Погрешность их значений равна 0.04% для n2,
0,02% для n1 и 0.002% для n3.
Энергии остальных возбужденных электронных состояний (Т0 < 20000 см‑1; 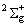 , 2Dg, 2Pu, и
, 2Dg, 2Pu, и 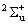 .) приняты по данным
[2001WAN/WAN].
.) приняты по данным
[2001WAN/WAN].
Термодинамические
функции CuF2(г) вычислялись по
уравнениям (1.3) - (1.6), (1.9), (1.10), (1.122) - (1.124),
(1.126), (1.129) и (1.168) - (1.170) в приближении "жесткий
ротатор – гармонический осциллятор" без учета различия молекулярных постоянных
для основного и возбужденных электронных состояний. Расчетная суммарная
погрешность для Fo(T) равна 0.95,. 2.4, 4.0 и 5.1 Дж×К‑1×моль‑1 при Т = 298.15,
1000,3000 и 6000 K соответственно.
Ранее термодинамические функции CuCl2(г) рассчитывались Брюэром с сотр. [63BRE/SOM] (Φ°(T) при Т = 298.15,
500, 1000, и 1500 K, а также H (298.15 K) - H (0)). Следующий набор молекулярных постоянных был выбран для расчета
авторами для линейной молекулы CuCl2:
r(Cu-Cl) = 2.09 Å,
n1 = 340
см‑1, n2 = 50
см‑1, и n3 = 496
см‑1. Энергии возбужденных электронных состояний оценены на
базе теории поля лигандов. Поскольку значение n2
вдвое занижено и энергии возбужденных электронных состояний занижены, значения
термодинамических функции, рассчитанные Брюэром и сотр. [63BRE/SOM] завышены.
Константа равновесия реакции CuCl2(г) = Cu(г) + 2Cl(г) вычислена по значению DrH°(0) = 611.747 ± 3.7 кДж×моль‑1, соответствующему принятым для СuCl2(к) энтальпиям
образования и сублимации. Этим величинам также соответствует значение
DfH°(СuCl2, г, 0) = -35.698 ± 3.1 кДж×моль‑1.
АВТОРЫ
Ежов Ю.С. ezhovyus@mail.ru
Гусаров А.В. a-gusarov@yandex.ru
Версия для печати

