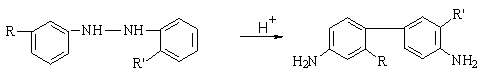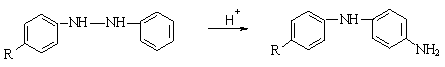|
Основные учебные курсы
для студентов Химического факультета МГУ
Органическая химия
Подробная программа лекций и
комментарии ко второй части курса
Подробная программа лекций и комментарии к второй
части общего курса лекций по органической химии (ППЛ) основаны на Программе общего курса органической химии,
разработанной на кафедре органической химии химического факультета МГУ. ППЛ
раскрывают наполнение второй части общего курса лекций фактическим материалом
по теории и практике органической химии. ППЛ предназначена в первую очередь для
студентов 3 курса, желающих хорошо и достаточно быстро подготовиться к
экзаменам и коллоквиумам и понять, какой объём знаний должен иметь студент для
получения отличной оценки на экзамене. ППЛ подготовлены таким образом, что
обязательный материал программы напечатан нормальным шрифтом, факультативный
материал – курсивом, хотя следует
признать, что такое деление иногда является достаточно условным.
Одной из целей данного пособия является помощь студентам правильно и
аккуратно составить конспект лекции, структурировать материал, сделать
правильные акценты в записи, отделить обязательный материал от второстепенного
при самостоятельной работе с конспектом или учебником. Необходимо отметить, что
несмотря на широчайшее распространение современных методов обучения и
доступность разнообразного учебного материала в учебниках и в Интернете, только
самостоятельная упорная, если не сказать тяжёлая работа по конспектированию
(лекций, учебников, других материалов), работа на семинарах, самостоятельное
написание важнейших уравнений и механизмов, и самостоятельное решение
синтетических задач способно привести к успеху в изучении органической химии
(да и других предметов). Авторы считают, что прослушивание курса лекций создаёт
основу для изучения органической химии и охватывает все темы, выносимые на
экзамен. Однако, прослушанные лекции, равно как и прочитанные учебники остаются
пассивным знанием до тех пор, пока не произойдёт закрепление материала на
семинарах, коллоквиумах, при написании тестов, контрольных работ и анализе
ошибок. В ППЛ отсутствуют уравнения химических реакций и механизмы важнейших
процессов. Этот материал доступен на лекциях и из учебников. Каждый студент
должен добывать какие-то знания самостоятельно: написать важнейшие реакции,
механизмы и лучше не один раз (самостоятельная работа с конспектом лекций, с
учебником, коллоквиум). Только то, что приобретается путём самостоятельной
кропотливой работы запоминается надолго и становится активным знанием. То, что
легко достаётся – легко теряется или забывается, причём это справедливо не
только по отношению к курсу органической химии.
Помимо материалов программного характера данная
разработка содержит ряд вспомогательных материалов, которые демонстрировались
на лекциях и которые необходимы, по мнению авторов, для лучшего понимания
органической химии. Эти вспомогательные материалы (цифры, таблицы и др.), даже
если они напечатаны нормальным шрифтом чаще всего не предназначены для
буквального запоминания, но нужны для оценки тенденций изменения свойств или
реакционной способности органических соединений. Поскольку демонстрируемые на
лекциях вспомогательные материалы, цифры, таблицы бывает трудно полностью и
качественно записать в конспект, размещение этих материалов в данной разработке
имеет целью помочь слушателям курса восполнить пробелы в записях и конспектах,
а на лекции сосредоточиться не на стенографировании цифр и таблиц, а на
восприятии и понимании материала, обсуждаемого лектором.
Авторы выражают искреннюю благодарность доценту М.В.Ливанцову
и доценту Ю.А.Вейцу за внимательный просмотр текста и ценные замечания.
АРОМАТИЧНОСТЬ.
1.
Алифатические (от греч. αλιφατικό-масло, жир) и ароматические (αρωματικόσ-благовоние)
соединения (ХIХ век).
2.
Открытие бензола
(Фарадей, 1825). Строение бензола (Кекуле, 1865). о-, м-, п-изомеры, орто-ксилол.
3. Другие
формулы, предложенные для бензола (Ладенбург, “Дьюар”, Тиле и др.). Изомеры
бензола (призман, бицикло[2,2,0]гекса-2,5-диен, бензвален, фульвен).
4. Метод молекулярных орбиталей
Хюккеля.
Независимое рассмотрение σ- и π- связей (т.е. образованных sp2 и
р-орбиталями). Молекулярные орбитали бензола (три орбитали связывающие: одна
орбиталь не имеет узлов, две орбитали имеют по одной узловой плоскости, все они
заняты, на них всего 6 электронов; три орбитали разрыхляющие. Две из них имеют 2
узловых плоскости, самая высокая по энергии разрыхляющая орбиталь имеет три
узловых плоскости. Разрыхляющие орбитали не заняты.
|
Ej(энергия
орбитали j уровня) = α+ mjβ
α –кулоновский интеграл, энергия С2р
орбитали,
β –
резонансный интеграл, энергия взаимодействия 2-х атомных орбиталей на
соседних атомах
mj = 2 сos(2jπ/N), где N- число атомов углерода в цикле.
|
Понятие о круге Фроста для бензола,
циклобутадиена и циклооктатетраена.
Правило Хюккеля. ПЛОСКИЕ,
МОНОЦИКЛИЧЕСКИЕ, СОПРЯЖЕННЫЕ углеводороды будут ароматическими, если цикл
содержит (4n+2) π – электронов.
Антиароматические
соединения. Неароматические соединения. Циклооктатетраен.
5.
Описание бензола по методу "валентных схем", теория резонанса (Полинг),
мезомерия, использование предельных структур.
6.
Аннулены. Метаноаннулены. Ароматические ионы. Конденсированные углеводороды.
Гетероциклы.
Несколько комментариев по
стабильности аннуленов.
[10]-аннулен – не плоский, не может быть ароматичным.
1,6-метано-[10]- аннулен - плоский,
(кроме мостика, разумеется!), он ароматичен.
[12]-аннулен
– неароматический полиен, стабилен ниже -70оС.
[14]-аннулены не плоские циклы, если нет 2-х мостиков.
Следовательно – не ароматичные.
[16]-аннулены
– обычные полиены.
[18]-аннулен – плоский, ароматичный. Знать особенность его ПМР спектра!
7. Подробное рассмотрение КРИТЕРИЕВ АРОМАТИЧНОСТИ.
|
Критерии ароматичности – квантовомеханический
– количество p-электронов
4n+2 (правило Хюккеля), см. комментарии ниже.
Энергетический( повышение термодинамической устойчивости за счет делокализации
электронов, так называемая энергия
делокализации – ЭД).
ЭД
в бензоле: (6a+8β) – (6a+6β) (для циклогексатриена) = 2β = 36 ккал/моль или 1,56 эВ – это ЭЭР (эмпирическая энергия резонанса).
Есть
еще несколько способов расчёта энергии резонанса: вертикальная энергия
резонанса (она же ЭД по Хюккелю, измеряют в единицах интеграла β, для бензола она равна 0,333), еще бывает (на 5++) ЭРД (т.е. энергия резонанса Дьюара, на 1
электрон, 0,145 эВ для бензола), еще бывает (на 5+++) ЭР по Гессу-Шааду, для
бензола: 0,065 эВ, то же, что и ЭДНОЭ в учебнике Реутов, Курц, Бутин. Еще
бывает (на 5++++) ТЭР (топологическая
ЭР). Еще "есть многое на свете, друг
Горацио, что и не снилось нашим мудрецам"
(В. Шекспир).
Энергетический
критерий из всех самый неудобный и неясный. Величины энергий для этого критерия
берут всегда расчетные, потому что, как правило, невозможно подобрать
соответствующую неароматическую молекулу для сравнения. Следует, поэтому,
спокойно относиться к тому, что существует множество различных оценок энергии
делокализации даже для классических ароматических молекул, а для более
сложных систем эти величины вообще отсутствуют. Никогда нельзя сравнивать
разные ароматические системы по величине энергий делокализации – нельзя
сделать вывод, что молекула А ароматичнее молекулы В, потому что энергия
делокализации больше.
Структурный – очень важный, если не самый важный, критерий, так
как имеет не теоретическую, а экспериментальную природу. Специфика геометрии молекул ароматических соединений заключается в
тенденции к копланарному
расположению атомов и выравниванию длин связей. У бензола выравнивание
длин связей идеально – все шесть С-С связей одинаковы по длине. У более
сложных молекул выравнивание не идеально, но значительно. В качестве критерия
берут меру относительного отклонения длин сопряженных связей от среднего
значения. Чем ближе к нулю, тем лучше. Эту величину можно проанализировать
всегда, если имеется структурная информация (экспериментальная или из
высококачественного квантовохимического расчета). Тенденция к копланарности
обуславливается выгодностью коллинеарности осей атомных р-орбиталей для их эффективного перекрывания. Возникает
вопрос: какое отклонение от плоскостного расположения допустимо без потери
ароматичности? Примеры искажения плоскости в ароматических молекулах даны на
лекции, их также можно найти в специальной литературе (см. ниже стр. 20).
Магнитный
(наличие кольцевого тока – диатропная система, влияние на химические сдвиги
протонов снаружи и внутри кольца, примеры – бензол и [18]-аннулен). Самый
удобный и доступный критерий, так как для его оценки достаточно спектра 1H ЯМР. Для точного определения используют
теоретические расчеты химических сдвигов.
Что такое диатропность?
Химический – склонность к реакциям замещения, а не
присоединения. Самый наглядный критерий, ясно различающий химию ароматических
соединений от химии полиенов. Но работает он далеко не всегда. В ионных
системах (например, в циклопентадиенил-анионе или тропилий-катионе) замещение
наблюдать невозможно. Реакции замещения иногда проходят и на неароматических
системах, а ароматические всегда в какой-то степени способны к реакциям
присоединения. Поэтому химический
критерий более правильно назвать ПРИЗНАКОМ ароматичности.
|
8. ПОНЯТИЕ АРОМАТИЧНОСТИ. ПРИЗНАКИ И КРИТЕРИИ
АРОМАТИЧНОСТИ. - Комментарии
Ароматичность– понятие, характеризующее совокупность особых структурных,
энергетических и магнитных свойств, а также особенностей реакционной
способности циклических структур с системой сопряженных связей.
Хотя ароматичность – одна из важнейших и наиболее
плодотворных концепций химии (не только органической), - не существует общепринятого краткого
определения этого понятия. Ароматичность понимается через совокупность
особых признаков (критериев), присущих ряду циклических сопряженных молекул в
той или иной мере. Часть этих критериев имеет экспериментальную, наблюдаемую
природу, но другая часть основывается на квантовой теории строения молекул. Ароматичность имеет квантовую природу.
Невозможно объяснить ароматичность с позиций классической структурной теории и
теории резонанса.
Не следует путать ароматичность с делокализацией и сопряжением.
В молекулах полиенов (1,3-бутадиена, 1,3,5-гексатриена и т.п.) проявляется явно
выраженная тенденция к делокализации электронов (см. 1-й семестр, химия диенов)
и образованию единой сопряженной электронной структуры, что проявляется в
спектрах (в первую очередь, электронных спектрах поглощения), некотором
изменении длин и порядков связей, энергетической стабилизации, особых
химических свойствах (электрофильное 1,4-присоединение в случае диенов и пр.). Делокализация
и сопряжение – необходимые, но не достаточные условия ароматичности. Можно дать
определение ароматичности как свойства, при котором сопряженное кольцо ненасыщенных связей проявляет бόльшую
стабильность, чем ту, которую можно было бы ожидать только при одном сопряжении. Однако этим определением нельзя пользоваться,
не имея экспериментальных или расчётных данных по стабильности циклической
сопряжённой молекулы.
Для того, чтобы молекула могла быть ароматической, она
должна содержать хотя бы один цикл, каждый из атомов которого располагает пригодной для образования
ароматической системы р-орбиталью.
Ароматическим в полном смысле этого слова считается (в случае выполнения
критериев, перечисленных ниже) именно этот цикл (кольцо, система колец).
В этом цикле
должно быть 4n+2 (то есть
2, 6, 10, 14, 18, 22 и т.п.) электронов.
Это правило называется правилом или критерием
ароматичности Хюккеля. Источник этого правила – сильно упрощенные
квантовохимические расчеты идеализированных циклических полиенов, произведенные
на заре развития квантовой химии. Дальнейшие исследования показали, что в
основе своей это простое правило дает верные предсказания ароматичности даже и
для очень сложных реальных систем.
Правилом, тем не
менее, нужно правильно пользоваться, иначе прогноз может быть неверен.
Общие рекомендации даны далее.
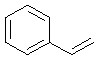
Молекула, содержащая хотя бы один ароматический цикл имеет право называться ароматической, но
этим обобщением не стоит злоупотреблять. Так, очевидно, что стирол содержит
бензольное кольцо, а следовательно может называться ароматической молекулой. Но
нас в стироле может интересовать и этиленовая двойная связь, никакого прямого
отношения к ароматичности не имеющая. С этой точки зрения стирол – типичный
олефин с сопряжённой двойной связью.
Никогда не забывайте, что химия есть наука
экспериментальная, и никакие теоретические рассуждения не заменяют и не
подменяют знание реальных свойств веществ. Теоретические концепции, даже столь
важные, как ароматичность, только помогают лучше понимать эти свойства и делать
полезные обобщения.
Какие
орбитали считаются пригодными для образования ароматической системы? – Любые орбитали, перпендикулярные плоскости цикла, и
а) принадлежащие входящим в цикл кратным (эндоциклическим
двойным или тройным) связям;
б) соответствующие неподеленным парам электронов у
гетероатомов (азота, кислорода, и т.п.) или карбанионов;
в) соответствующие шестиэлектронным (секстетным)
центрам, в частности карбокатионам.
Обратите внимание, что перечисленные фрагменты а), б),
в) дают четное число электронов в общую систему: любые кратные связи – 2
электрона, неподеленные пары – 2 электрона, вакантные орбитали – 0 электронов.
Что не
годится или не вносит вклад в ароматическую систему:
а) ониевые
формы катионных центров – то есть
катионы, содержащие полный октет электронов. При этом такой центр разрывает сопряженную
систему, например, N-метилпиррол ароматичен (6
электронов в цикле), а N,N-диметилпирролий неароматичен (аммонийный азот не
вносит вклад в π-систему):
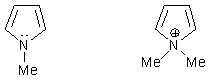
Внимание – если ониевый центр является частью кратной
связи, то именно кратная связь участвует в образовании ароматической системы,
поэтому, например, ароматичен N-метилпиридиний
(6 π-электронов, по два от каждой из трех кратных связей).
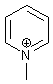
Очень большое подспорье в рассмотрении аналогичных
систем дает концепция изоэлектронности. Изоэлектронные
системы обычно аналогичны и в смысле ароматичности. В этом смысле, например, N-метилпиридиний изоэлектронен метилбензолу. Оба,
очевидно, ароматичны.
б) неподеленные
пары, лежащие в плоскости кольца. На одном атоме только одна π-орбиталь
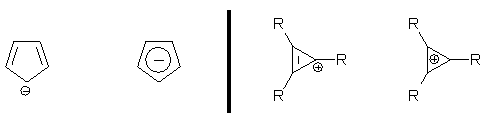
может вносить вклад в ароматическую систему. Поэтому, в циклопентадиенил-анионе
карбанионный центр дает вклад в 2 электрона, а в фенил-анионе атом углерода
карбанионного центра дает вклад в 1 электрон, как и в молекуле бензола.
Фенил-анион изоэлектронен пиридину, циклопентадиенил-анион – пирролу.
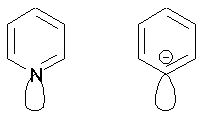
Все
ароматичны.

в) Экзоциклическая двойная связь или радикальный
центр. Такие структуры, как правило, неароматичны, хотя каждая такая структура нуждается в особом рассмотрении с
привлечением реальных экспериментальных данных.
Например, хиноны неароматичны, хотя а) обладают
плоскими полностью сопряженными циклами, содержащими 6 электронов (четыре от
двух кратных связей в цикле плюс два от двух экзоциклических связей).
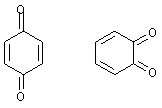
Наличие в некоторой сопряженной структуре так
называемых хиноидных фрагментов, то есть систем связей с двумя экзоциклическими
двойными связями, всегда является источником нестабильности, и благоприятствует
процессам, переводящим систему с хиноидным фрагментом в нормальную
ароматическую систему. Так, антрацен является 14-электронной ароматической
системой, содержащей хиноидный фрагмент, поэтому, антрацен легко присоединяет
бром или диенофилы, так как в продуктах уже два полноценных ароматических
бензольных кольца:
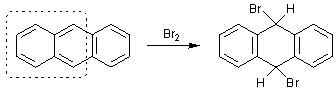
Ароматичность полициклических структур представляет собой достаточно сложную теоретическую
задачу. С формальной точки зрения, если в системе есть хотя бы одно бензольное
кольцо, то ее можно считать ароматической. Такой подход, однако, не дает возможности
рассмотреть свойства молекулы в целом.
Современный подход к полициклическим системам состоит
в том, чтобы найти в них все
возможные ароматические подсистемы, начиная
с максимально большого – внешнего контура. В этом смысле, например,
нафталин можно представить в виде общей 10-электронной системы (внешний контур)
и двух одинаковых 6-тиэлектронных бензольных колец.
Если внешний контур не ароматичен, то следует искать
меньшие ароматические контуры. Так, например, дифенилен по внешнему контуру
имеет 12 электронов, что не соответствует правилу Хюккеля. Однако мы легко
найдем в этом соединении два практически независимых бензольных кольца.
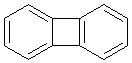
Если
бициклические углеводороды плоские и имеют сопряженные двойные связи, правило
Хюккеля работает для би- и полициклических углеводородов, имеющих одну общую
связь (нафталин, антрацен, фенантрен,
и т.п., а также азулен). Правило Хюккеля плохо работает для
конденсированных циклов, имеющих атом углерода, общий для 3 циклов. Правило
подсчета пар электронов методом "обхода по периметру, или по одному из контуров"
может помочь в этом случае, например:
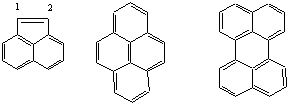
аценафтилен пирен перилен
сумма π-электронов: 12 16 20
в том числе по
периметру, 10 14 18 (по контуру нафталина
– 10 и 10)
Однако,
для подобных сложных циклов и это правило не всегда может работать. Более того,
оно ничего не говорит о реальных свойствах молекулы. Например, аценафтилен, имеет
обычную двойную связь между атомами 1 и
2 .
Различные примеры изоэлектронных ароматических
гетероциклов.
ПИРРОЛ – ФУРАН – ТИОФЕН (6 π—электронов).
ПИРИДИН – ПИРИДИНИЙ –
ПИРИЛИЙ (6 π—электронов).
Пиридазин
– ПИРИМИДИН – пиразин (6 π—электронов).
Оксазолы
– тиазолы – ИМИДАЗОЛ (6 π—электронов).
ИНДОЛ
– ХИНОЛИН (10 π—электронов).
О "гайках" .
В учебной литературе ароматические циклы часто обозначают с помощью окружности внутри
многоугольника. Со всей определенностью отметим, что такой способ обозначения
следует избегать во всех случаях, когда только возможно. Почему?
Потому что:
а) в сложных полициклических структурах кружки не
имеют определенного смысла и не позволяют понять, где живет ароматичность – в
отдельных циклах или в целом. Если нарисовать "гайками", например, антрацен, то
не будет понятно, в чем кроется причина его "не-совсем-ароматичности" и
ярко-выраженных диеновых свойств
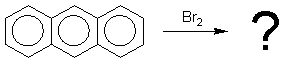
б) даже самые классические ароматические системы типа
бензола и его производных могут проявлять неароматические полиеновые свойства,
для рассмотрения которых необходимо видеть структуру кратных связей.
в) именно кекулевская структура необходима для рассмотрения
эффектов заместителей с помощью незаменимого инструмента – резонансных структур.
"Гайка" в этом отношении совершенно бесплодна. Так, пользуясь формулой Кекуле,
мы отлично поймем причину высокой кислотности п-нитрофенола и ярко-желтый цвет п-нитрофенолята. А что будем делать с "гайкой"?
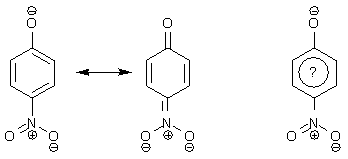
Предпочтительным является простой
"кекулевско-бутлеровский" способ, соответствующий классической теории строения
и в явном виде обозначающий кратные связи. Нарисовав такую классическую
структуру, вы всегда можете рассуждать о ее ароматичности или неароматичности,
используя соответствующие правила и критерии. Именно классическая кекулевская
структура принята как стандарт во всех ведущих международных химических
журналах.
А когда все же уместны кружки? Для обозначения небензоидных ароматических систем,
особенно заряженных. В этом случае классическое обозначение несколько неуклюже
и не показывает делокализацию заряда.
Также без кружков трудно обойтись в металлоорганической
химии, где ароматические системы часто играют роль лигандов. Попробуйте без
кружков отразить структуру ферроцена или других комплексов, содержащих
циклопентадиенильный лиганд!

Плоскостность.Цикл,
претендующий на звание ароматического и содержащий нужную непрерывную систему p-орбиталей должен быть плоским (или почти плоским). Это требование – одно из самых
неприятных, так как определить "на глазок", какой цикл является плоским, а
какой нет, весьма не просто. В качестве простых подсказок можно рассмотреть
следующие положения:
а) циклические сопряженные системы, содержащие 2 или 6
электронов и удовлетворяющие рассмотренным условиям, как правило, плоские и
ароматические. Такие системы обычно реализуются в циклах малого и среднего
размера (2-8 членных);
б) циклические ионные системы с числом электронов 2,
6, 10, 14, практически обязательно ароматичны, так как ароматичность и является
причиной существования и устойчивости таких ионов;
в) нейтральные системы с 10, 14, 18 и более электронами в одном единственном цикле
большого размера, напротив, практически всегда нуждаются в дополнительных мерах
по стабилизации плоской структуры в виде дополнительных мостиков, так как
выигрыш энергии за счет образования большой ароматической системы не
компенсирует ни энергии напряжения, возникающей в макроциклах, ни энтропии,
теряемой при образовании единственной плоской структуры.
Внимание: Чтение следующего параграфа
категорически не рекомендуется лицам со слабыми и неустойчивыми знаниями. Все,
кто имеет рейтинг менее 99 баллов, могут
пропустить этот параграф.
Антиароматичность. Системы, удовлетворяющие всем условиям,
рассмотренным выше (плоские циклы с непрерывной системой π-орбиталей), но
числом электронов 4n, считаются антиароматическими – то есть реально несуществующими. Но если
в случае ароматичности мы имеем дело с реальными молекулами, то в случае
антиароматичности проблема сложнее. Важно понять, что настоящая
антиароматическая система находится не в минимуме, а в максимуме потенциальной
энергии, то есть представляет собой не молекулу, а переходное состояние.
Антиароматичность – чисто теоретическая концепция, описывающая, почему
некоторые циклические сопряженные системы или совсем неустойчивы и не могли
быть получены даже ценой огромных усилий, или проявляют явные тенденции к
существованию в виде обычного полиена с чередующимися простыми и кратными
связями.
Например, циклобутадиен был бы антиароматичен,
если бы существовал в виде квадратной молекулы со связями одинаковой длины. Но
такой квадратной молекулы в Природе нет. Поэтому корректно следует сказать так:
гипотетический квадратный циклобутадиен
антиароматичен, и поэтому не существует. Экспериментально, при очень
низких температурах выделяли замещенные циклобутадиены, но они по структуре
оказывались типичными неароматическими диенами – имели ясное различие между
короткими двойными и длинными простыми связями.
Реально существующие плоские сопряженные молекулы с 4n электронами всегда представляют собой
высокореакционноспособные неароматические полиены. Например, бензоциклобутадиен
реально существует (8 электронов во внешнем контуре), но обладает свойствами
чрезвычайно активного диена.
Антиароматичность
– исключительно важное понятие в
теории ароматичности. Теория ароматичности предсказывает как существование
особо стабильных ароматических систем, так и нестабильность антиароматических
систем. Оба этих полюса важны.
Антиароматичность
– очень важное понятие в химии. Все ненасыщенные сопряженные циклические
системы, содержащие антиароматическое число π-электронов, всегда обладают
очень высокой реакционной способностью в различных реакциях присоединения.
9. Тривиальные примеры синтеза
небензоидных ароматических ионов.
Циклопропенилий-катион,
тропилий-катион
Циклопентадиенилид-анион.
Ароматические карбоциклические анионы С8, С10, С14.
10. Факультативно: попытки синтеза
антиароматических молекул – циклобутадиен, циклопентадиенилий-катион.
Развитие понятия ароматичности. Циклобутадиенжелезо трикарбонил. Объемная,
сферическая ароматичность,
гомоароматичность и др.
11. Получение ароматических
углеводородов.
1.
Промышленные источники – нефть и каменный уголь.
Риформинг. Цепочка: гептан – толуол – бензол –
циклогексан.
2.
Лабораторные способы:
а) реакция Вюрца-Фиттига (устаревший способ, имеющий
скорее историческое значение, не следует применять при решении
задач),
б) каталитическая тримеризация ацетилена,
в) кислотно-катализируемая тримеризация ацетона и др.
кетонов;
г) кросс-сочетание, как некаталитическое с
использованием купратов, так и каталитическое в присутствии комплексов палладия,
д) реакция Фриделя-Крафтса, в основном следует
использовать ацилирование с восстановлением по Клемменсену (алкиларилкетоны)
или Кижнеру-Вольфу (любые кетоны и альдегиды),
е) ароматизация любых производных циклогексана,
циклогексена, циклогексадиена под действием серы (сплавление, годится только
для простейших соединений) или дихлордицианбензохинона (ДДХ или DDQ, реагент общего назначения).
12. Свойства кольца и алифатической
боковой цепи в ароматических углеводородах.
1.
Гидрирование. Когда
бывает частичное гидрирование колец? Гидрирование функциональных групп (С=С,
С=О) без гидрирования кольца. Примеры.
2.
Восстановление по Бёрчу
(Na, жидк. NH3). Зачем нужен EtOH? Влияние доноров и акцепторов в кольце на направление
реакции.
3.
Свободнорадикальное
галогенирование бензола (было в школе!). Галогенирование толуола и его
гомологов в боковую цепь. Избирательность галогенирования.
4.
Окисление боковой цепи и
поликонденсированных ароматических углеводородов. Озонирование бензола и других ароматических соединений.
5.
Реакция Дильса-Альдера
для бензола и антрацена. Условия.
6.
Реакция щелочных металлов и Mg с нафталином
и антраценом (факультативно).
ЭЛЕКТРОФИЛЬНОЕ ЗАМЕЩЕНИЕ В АРОМАТИЧЕСКОМ
РЯДУ.
1.
Почему именно
электрофильное замещение (ЭЗ)?
2.
Какие бывают электрофилы
и какие реакции ЭЗ мы будем разбирать подробно? (протонирование, нитрование,
сульфирование, галогенирование, алкилирование, ацилирование, формилирование). Через месяц будут рассмотрены: азосочетание,
нитрозирование, карбоксилирование).
3.
Упрощенный механизм
электрофильного замещения в ароматическом кольце (без π-комплексов).
Аренониевые ионы. Сходство с аллильным катионом. Изображение аренониевых ионов
на бумаге – резонансные структуры или "подкова" – обязательно научитесь
рисовать резонансные структуры для s-комплексов, так как "подкова" заведет в тупик, когда
дойдем до влияния заместителей на направление электрофильного замещения. Протонирование
аренов.
4.
Доказательства
существования π-комплексов на примере реакции DCl и бензола (Г. Браун 1952). Доказательства
существования σ-комплексов.
5.
Обобщенный механизм ЭЗ,
включающий образование π- и σ-комплексов. Лимитирующая стадия при ЭЗ
в бензольном кольце. Понятие о
кинетическом изотопном эффекте. Еще раз вспомним, что такое переходное
состояние и интермедиат.
6.
Ориентация при
электрофильном замещении: орто-, мета,
пара-, ипсо. Ориентанты первого и второго рода. Обязательно рисовать
резонансные структуры для s-комплексов с различными заместителями. Отдельно разберите влияние на
структуру s-комплексов заместителей
с индуктивными и мезомерными эффектами, а
также сочетанием разнонаправленных эффектов. Факторы парциальной
скорости. Согласованная и несогласованная ориентация. Примеры различного
соотношения о-/п- изомеров в случаях, когда в кольце имеется заместитель 1-го
рода (например, пространственно-затруднённый) или 2-ого рода (орто-эффект). ЯМР
бензолониевых ионов и некоторых аренов.
7.
Рассмотрение конкретных
реакций электрофильного замещения.
Нитрование. Агенты. Экзотические
агенты. Атакующая частица. Особенности нитрования разных классов соединений
– нитроаренов (условия), галогенбензолов (деление о- и п- изомеров. Каким
образом?), нафталина и дифенила.
Нитрование ароматических аминов (защитные группы, как сделать о- и п-
изомеры? Можно ли нитровать анилины в м-положение?). Нитрование фенола
(условия, деление о- и п- изомеров).
7. Сульфирование аренов. Агенты,
природа электрофила, обратимость. Особенности сульфирования нафталина, толуола,
фенола, анилина, защита сульфогруппой при реакциях ЭЗ.
8. Производные сульфокислот:
тозилхлорид, тозилаты, сульфамиды. Восстановление сульфогруппы.
9. Галогенирование. Ряд
галогенирующих агентов по убыванию активности (знать хотя бы 3 примера). Природа
электрофила, особенности галогенирования толуола, галогенбензолов, уметь
получать все галогеннитробензолы, галогенирование нафталина, бифенила, анилина,
фенола, анизола. Особенности иодирования. Хлорирование иодбензола без
электрофильных катализаторов. Соединения поливалентного иода (PhICl2, PhI=O, PhI(OAc)2)
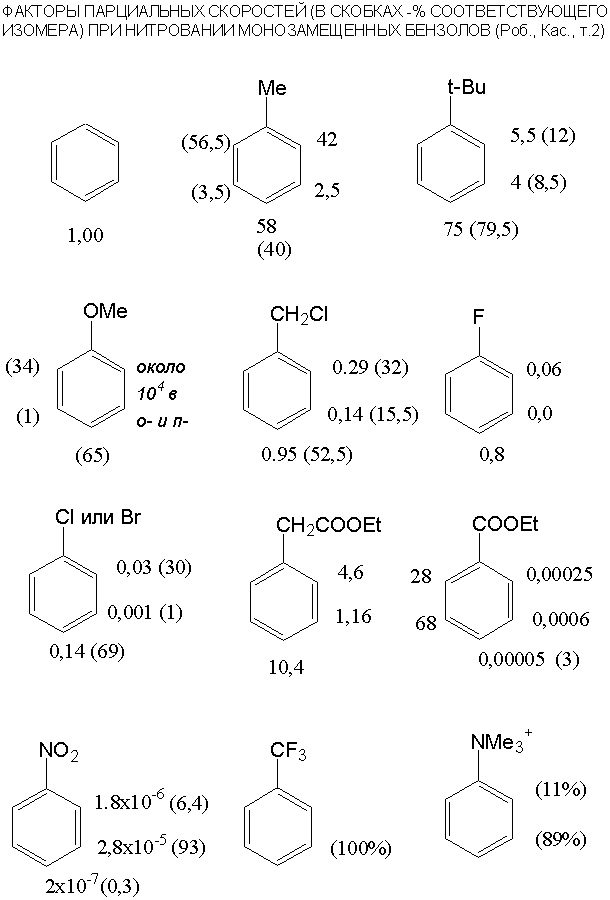
10.Алкилирование
и ацилирование по Фриделю-Крафтсу. Алкилирование– 3 недостатка, примеры
синтезов, обратимость, влияние галогена
в RHal, агенты, внутримолекулярное алкилирование,
ограничения по заместителям, особенности алкилирования фенолов и аминов, синтез
н-алкилбензолов. Ацилирование – сравнение с алкилированием, реагенты, циклические ангидриды в ацилировании,
внутримолекулярные реакции, перегруппировка Фриса.
Таблица
1.
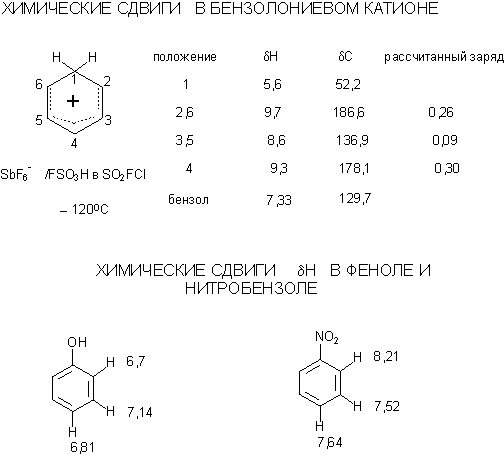
Таблица 2. Данные по нитрованию галогенбензолов.
|
Соединение
|
продукты, %*
|
относительная
скорость
нитрования
(бензол =1)**
|
Фактор
парциальной скорости для о- и п-
положения (бензол = 1)
|
|
|
орто
|
мета
|
пара
|
|
|
|
С6Н5
– F
|
12
|
|
87
|
0,15
|
0,054 (о)
0,783 (п)
|
|
С6Н5
– Cl
|
30
|
0,9
|
69
|
0,033
|
0,030 (о) 0,136(п)
|
|
С6Н5
– Br
|
37
|
1,2
|
62
|
0,030
|
0,033 (о) 0,116(п)
|
|
С6Н5
– I***
|
38
|
1,8
|
60
|
0,18
|
0,205 (о) 0,648(п)
|
*) К. Ингольд.
Теоретические основы органической химию М., "Мир", 1973, с. 263;
**) там же с. 247;
***) согласно новейшим исследованиям, механизм электрофильного замещения в
арилиодидах может быть более сложным, чем было принято ранее.
О разделение о- и
п- изомеров дизамещенных аренов кристаллизацией.
Таблица 3. Т. пл. о- и
п-изомеров дизамещенных аренов в оС.
|
Заместители
|
Т.
пл. о- и п-изомеров
|
Заместители
|
Т. пл. о- и п-изомеров
|
|
Ме, NO2
|
9, 54
|
Ме, С1
|
-34,
7,5
|
|
С1, NO2
|
33, 84
|
Ме, Br
|
-28
28,5
|
|
Br,
NO2
|
43, 127
|
OH, C1
|
7,
37
|
|
Ме, NH2
|
-16, 45
|
OH, Br
|
5,6,
63,5
|
СРАВНЕНИЕ РЕАКЦИЙ АЛКИЛИРОВАНИЯ И
АЦИЛИРОВАНИЯ ПО ФРИДЕЛЮ-КРАФТСУ.
|
|
АЛКИЛИРОВАНИЕ
|
АЦИЛИРОВАНИЕ
|
|
РЕАГЕНТ
|
AlkHal, AlkOH, алкены.
(Нельзя ArHal!).
|
Галогенангидриды карбоновых кислот
(КК), ангидриды КК, редко – КК
|
|
КАТАЛИЗАТОР
|
Кислоты Льюиса, особенно б/в галогениды Аl, Fe, Sn и др., BF3, H2SO4, H3PO4,
катиониты.
|
AlCl3 (не
менее моля на моль, лучше еще больше), H2SO4, H3PO4.
|
|
ПРОДУКТ
|
Алкил- и полиалкиларены.
|
Ароматические кетоны. Можно ввести только одну
ацильную группу.
|
|
ОСОБЕННОСТИ
И НЕДОСТАТКИ
|
Практически малопригодна из-за множества побочных
реакций, а именно:
1) полиалкилирование,
2)
изомеризация исходных н-алкилов во втор.- и трет- алкилы.
3)
изомеризация полиалкилбензолов в смесь или в более стабильный продукт.
|
Очень удобная реакция, практически не осложненная
побочными реакциями. Образуется, как правило, только пара-изомер. Если п-положение занято, то орто-изомер (по
отношению к наиболее сильному ориентанту).
|
|
ОБРАТИМОСТЬ
|
ЕСТЬ. (см.
ниже)
|
НЕТ.
|
|
ОБЛАСТЬ
ПРИМЕНЕНИЯ
|
НЕЛЬЗЯ ИСПОЛЬЗОВАТЬ для аренов, содержащих заместители
II-го рода. Можно использовать для арилгалогенидов.
|
|
ОСОБЕННОСТИ
ПРИМЕНЕНИЯ К ФЕНОЛАМ
|
НЕЖЕЛАТЕЛЬНО использовать AlCl3.
МОЖНО использовать катализаторы – H3PO4, HF со спиртами
в качестве алкилирующих реагентов.
|
CAcClможет идти
ацилирование по кислороду. При нагревании эфира фенола идёт перегруппировка ФРИСА (кат. – AlCl3).
Иногда для реакции Фр-Кр можно использовать АсОН\ВF3
Синтез фенолфталеина.
|
|
ОСОБЕННОСТИ
ПРИМЕНЕНИЯ К АРОМАТИ-
ЧЕСКИМ,
АМИНАМ
|
Прямое
алкилирование практически невозможно, так как нельзя использовать AlCl3, H2SO4, H3PO4, HF (атака AlCl3
или Н+ или алкила по
азоту – в результате электронодонор-ные свойства азота уменьша-ются. При действии RHalобразуются N-алкиланилины).
|
Идет ацилирование по азоту. Катализаторы образуют
комплексы по азоту. Возможно ацилирование при использовании двух экв.
ацилирующего агента и ZnCl2с
образованием п-ацил-N-ациланилинов.
|
|
|
|
|
Примечание:
Обратимость реакции алкилирования по Фриделю-Крафтсу
приводит к тому, что в системе одновременно идут все возможные реакции алкилирования
и деалкилирования, причем затрагивается и мета-положение, так как алкильная –
группа активирует все положения
бензольного кольца, хотя и в разной степени.
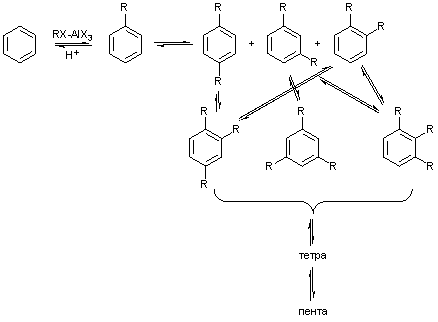
Тем не менее, из-за преимущественной орто-пара-ориентации
процессов алкилирования и обратного деалкилирования под действием электрофила,
например, при ипсо-атаке протона, в смеси при продолжительной реакции накапливаются наименее реакционноспособные и
более термодинамически стабильные 1,3- и 1,3,5-изомеры, так как в них алкилы хуже ориентируют атаку
протона под другие алкилы:
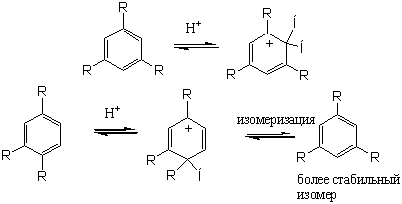
Похожие
причины определяют и образование разных региоизомеров при сульфировании, с той
существенной разницей, что сульфогруппа есть ориентант второго рода, что
затрудняет полисульфирование.
12.
ФОРМИЛИРОВАНИЕ – введение СНО группы.
Формилирование – частный случай ацилирования.
Многие
производные муравьиной кислоты могут формилировать арены. Реакции
формилирования с СО, НСN, HCO(NMe2)2. Специфика подбора электрофильных
катализаторов для реакций формилирования.
ГАТТЕРМАН-КОХ (1897) – ArH + CO + HCl (AlCl3/ Cu2Cl2). Бывает ли НС(О)С1? А НС(О)F?
ГАТТЕРМАН – НСN б\в + НС1
газ. Кат. AlCl3 или ZnCl2.
Гаттерман-Адамс
(факультативно) – Zn(CN)2 + HCl. Можно
использовать 1,3.5. триазин,/ НС1/А1С13 (факультативно), или С12СНОR (на 5+++)
Губен-Геш
(ацилирование с RCN, HCl и ZnCl2).
ФОРМИЛИРОВАНИЕ ПО ВИЛЬСМЕЙЕРУ-ХААКУ. Только
электронообогащенный арен! + ДМФА + РОС13 (можно SOCl2, COCl2).
13.
Реакция гидроксиметилирования, конденсация карбонильных соединений с аренами
(ДДТ, дифенилолпропан), хлорметилирование.
14.Применимость реакций формилирования и гидроксиметилирования.
Гаттерман-Кох - алкилбензолы, бензол,
галогенбензолы.
Гаттерман – активированные арены,
толуол.
Вильсмейер-Хаак – только активированные
арены
Хлорметилирование – фенол, анизол,
алкил- и галогенбензолы.
Гидроксиметилирование – активированные арены.
(Активированные
арены – это анилины, фенол и эфиры фенола.)
15. Триарилметановые красители. Кристаллвиолет (4-Ме2N-C6H4)3С+Х-.
Синтез из п-Ме2N-C6H4CHO + 2 Ме2NPh + ZnCl2 → ЛЕЙКО-ФОРМА (белый цвет). Далее окисление (PbO2 или
другой окислитель) в трет-спирт,
затем обработка кислотой, появление окраски.
ФАКУЛЬТАТИВНЫЙ МАТЕРИАЛ .
1) Меркурирование
бензола с Hg(OAc)2 Гексамеркурирование бензола с Hg(OAcF)2. Получение гексаиодбензола.
2) Декарбоксилирование ароматических
кислот ArCOOH (нагревание с порошком меди в хинолине) = ArH + CO2. Если есть электроноакцепторные группы в кольце, то
можно просто сильно нагреть соль аренкарбоновой кислоты. Если есть доноры,
особенно в орто- положении, возможно замещение протоном карбоксильной группы,
но это редко!
3)
Экзотические электрофилы в реакциях с аренами: (HN3/AlCl3 – дает анилин), R2NCl/ AlCl3 дает R2NAr) (SCl2/AlCl3 дает Ar2S.
Роданирование анилина или фенола дироданом (SCN)2.
Образование 2-аминобензотиазолов.
4)
Существует большое количество "хитрых" реакций,
которые запомнить невозможно, и не нужно, например PhOH + TlOAc + I2 = о-иодфенол, или PhOH + t-BuNH2 + Br2, -70oC =
о-бромфенол
НУКЛЕОФИЛЬНОЕ ЗАМЕЩЕНИЕ В
АРОМАТИЧЕСКОМ РЯДУ.
Почему нуклеофильное
замещение в аренах, не содержащих сильных электроноакцепторных групп,
происходит с большим трудом?
1.
SNAr –
ПРИСОЕДИНЕНИЕ-ОТЩЕПЛЕНИЕ.
1) Природа интермедиата. Комплексы
Мейзенгеймера. (Условия стабилизации интермедиата.) ЯМР 13С, м.д.:
3(ипсо), 75.8(о), 131.8(м), 78,0(п).
2)
Нуклеофилы. Растворители.
3)
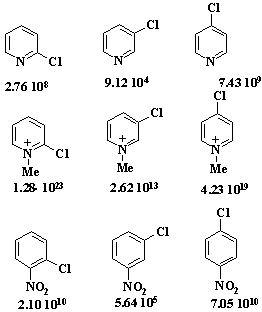
Ряд подвижности галогенов. F (400)>>NO2(8)>Cl(1) ≈ Br(1.18)>I (0.26). Лимитирующая стадия.
4)
Ряд активирующей способности заместителей (в каком положении?) NO2(1)>MeSO2(0.05)>CN(0.03)>Ac(0.01).
5)
Примеры конкретных реакций и конкретных условий.
6)
Факультативно: возможность замещения NO2- группы. Избирательное замещение NO2- группы. Пространственные факторы.
7) Нуклеофильное замещение водорода в ди- и
тринитробензоле. Зачем нужен окислитель?
2.
АРИНОВЫЙ механизм – (ОТЩЕПЛЕНИЕ-ПРИСОЕДИНЕНИЕ).
Меченый
хлорбензол и орто-хлортолуол, амиды калия или натрия в жидком NH3 .
Механизм.
Гидролиз
о-, м-, и п-хлортолуола, NaOH, H2O, 350-400oC, 300 атм.
ОЧЕНЬ ЖЕСТКИЕ УСЛОВИЯ!
Важность
индуктивного эффекта. Случай о-хлоранизола.
Медленная
стадия – отрыв протона (если Hal=Br, I) или отрыв
галогенид- аниона (если Hal=Cl, F).Отсюда – необычный ряд подвижности
галогенов: Br>I>Cl>F
Способы
генерирования дегидробензола. Строение дегидробензола – в этой частице нет
тройной связи! Улавливание дегидробензола.
3.
Механизм SRN1. Довольно
редкий механизм. Генерация анион-радикалов – электрический ток, или облучение,
или металлический калий в жидком
аммиаке. Реакционная способность ArI>ArBr. Несколько примеров. Какие нуклеофилы можно
использовать? Применение SRN1: реакции для a-арилирования карбонильных соединений через еноляты.
4.
Нуклеофильное замещение в присутствии меди. Синтез дифенилового эфира, трифениламина, гидролиз о-хлоранизола.
5.
Несколько
редких примеров. Синтез салициловой к-ты из бензойной, нуклеофильное замещение
в гексафторбензоле.
6.
SN1 Ar – см. тему "Диазосоединения".
Дополнительная литература по теме
"Ароматические соединения"
М.В.Горелик, Л.С.Эфрос. Основы химии и
технологии ароматических соединений. М., "Химия", 1992.
НИТРОСОЕДИНЕНИЯ.
Минимум знаний по алифатическим
нитросоединениям.
1.
СИНТЕЗ:
а) прямое нитрование в газовой фазе – только простейшие (1 семестр, тема –
алканы).
б) RBr + AgNO2 (эфир) = RNO2
(I) + RONO (II). Соотношение I и II зависит от R: Rперв. 80:10; Rвтор. 15:30.
Rтрет 0 : 10
: 60 (Е2, алкен). Можно использовать NaNO2 в ДМФ. Тогда количество RNO2 больше даже для вторичных R. Способ б)
хорош для RX, активных в SN2-замещении, например СlСН2COONa + NaNO2 в воде при 85оС. (тема: нуклеофильное
замещение и амбидентные анионы, 1 семестр).
в) НОВЫЙ
СПОСОБ СИНТЕЗА – окисление аминогруппы с CF3CO3H(из (CF3CO)2О + H2O2 в CH2Cl2 или MeCN).Годится
для алифатических и ароматических аминов. Иногда
можно брать м-ХНБК (м-хлорнадбензойная кислота, m-CPBA, продажный реагент). НЕ БРАТЬ ДЛЯ ОКИСЛЕНИЯ KMnO4 или K2Cr2O7!
Особенно для ароматических аминов!
2.
СВОЙСТВА. Самое главное свойство – высокая СН-кислотность, таутомерия нитро- и аци-формы (рКа МеNO2 10,5). Равновесие устанавливается медленно! С NaOH реагируют обе формы, а с содой – только аци-форма!
(Ганч).
Высокая СН-кислотность делает нитросоединения
аналогами енолизуемых карбонильных соединений. Кислотность нитрометана близка к
кислотности ацетилацетона, а не простых альдегидов и кетонов, поэтому
используются достаточно слабые основания – щелочи, карбонаты щелочных металлов,
амины.
Реакция Анри (Генри) – аналог альдольной или
кротоновой конденсации. Так как реакция Анри проводится в мягких условиях,
продуктом часто является нитроспирт (аналог альдоля), а не нитроолефин (аналог
кротонового продукта). RСН2NO2 – всегда
CH-компонента!
Реакции Михаэля и Манниха для RNO2. Факультативно: галогенирование в NaOH, нитрозирование, алкилирование анионов.
ВОССТАНОВЛЕНИЕ АРОМАТИЧЕСКИХ
СОЕДИНЕНИЙ.
1) Важнейшие промежуточные
продукты восстановления нитробензола в кислой среде (нитрозобензол,
фенилгидроксиламин) и щелочной среде (азоксибензол, азобензол, гидразобензол).
2) Селективное восстановление
одной из нитрогрупп в динитробензоле.
3) ВАЖНЕЙШИЕ СВОЙСТВА
ПРОДУКТОВ НЕПОЛНОГО ВОССТАНОВЛЕНИЯ НИТРОАРЕНОВ.
3а) Бензидиновая перегруппировка (Б.П.).
ВЫХОД
85% для бензидина. (R, R’ = H или др.
заместитель). ОБРАТИТЬ ВНИМАНИЕ НА
ПОЛОЖЕНИЕ R и R’ до и после перегруппировки!
Еще
15% - побочные продукты – в основном дифенилин (2,4’-диаминодифенил) и орто-бензидин.
Кинетическое
уравнение: V=k[гидразобензол][H+]2 – как правило, необходимо протонирование по обоим
атомам азота.
Бензидиновая перегруппировка – внутримолекулярная
реакция. Доказательство. Механизм - согласованная [5,5]-сигматропная
перегруппировка. Согласованный
процесс для бензидина.
Если
одно или оба пара-положения исходных гидразобензолов занято (R=Hal. Alk, AlkO, NH2, NMe2), может
происходить семидиновая перегруппировка с образованием СЕМИДИНОВ.
Некоторые заместители, например SO3H, CO2H, RC(O), находящиеся
в п-положении, могут отщепляться с образованием продуктов обычной Б.П.
Б.П. используется в производстве
азокрасителей, диаминов, напр. бензидина, толидина, дианизидина. Открыта
Н.Н.Зининым в 1845 г.
БЕНЗИДИН – КАНЦЕРОГЕН.
4) АЗОБЕНЗОЛ Ph-N=N-Ph. Син- анти-
изомерия.
АЗОКСИБЕНЗОЛ Ph-N+(→О-)=N-Ph. (Задача:
синтез несимметричных азо- и азоксибензолов из нитрозоаренов и ароматических
аминов или арилгидроксиламинов соответственно, или синтез азоксибензолов из
нитробензолов и ароматических аминов (NaOH, 175oC).
5) ФЕНИЛГИДРОКСИЛАМИН. Перегруппировка в кислой среде.
На
5+: родственные перегруппировки:
N-нитрозо-N-метиланилин (25oС), N-нитроанилин (10oС, было), Ph-NH-NH2 (180oC). Механизм обычно межмолекулярный.
6)
НИТРОЗОБЕНЗОЛ и его димер.
О реакции нитробензола RMgX с образованием алкилнитрозобензолов и других
продуктов. Эта реакция показывает, почему НЕЛЬЗЯ ДЕЛАТЬ реактивы Гриньяра из галогеннитробензолов!
МЕТОДЫ ПОЛУЧЕНИЯ АМИНОВ,
известные из материалов предыдущих лекций.
1.
Алкилирование аммиака и
аминов по Гофману
2.
Восстановление нитрилов,
амидов, азидов, оксимов.
3.
Восстановление
ароматических нитросоединений.
4.
Перегруппировки Гофмана,
Курциуса и Шмидта.
5.
(Гидролиз амидов.)
Новые способы.
1.
Восстановительное аминирование С=О (каталитическое).
2.
Реакция Лейкарта (Эшвайлера-Кларка).
3.
Синтез Габриэля,
4.
Реакция Риттера.
5. Каталитическое арилирование аминов в
присутствии медных и палладиевых катализаторов (реакции Ульмана, Бухвальда-Хартвига)
– наиболее мощный современный метод синтеза разнообразных аминов.
Химические свойства аминов, известные из предыдущих лекций.
1.
Нуклеофильное замещение
(алкилирование, ацилирование).
2.
Нуклеофильное
присоединение к С=О (имины и енамины).
3.
Элиминирование по
Гофману и по Коупу (из окисей аминов).
4.
Реакции электрофильного
замещения в ароматических аминах.
5. Основность аминов (школьная программа).
Новые свойства.
1.
Основность аминов (новый
уровень знаний). Что такое рКа и рКb.
2.
Реакция с азотистой
кислотой.
3.
Окисление аминов.
4. Разное – проба
Хинсберга, галогенирование аминов.
ДИАЗОСОЕДИНЕНИЯ.
1.
ДИАЗО- и АЗО-
соединения. СОЛИ ДИАЗОНИЯ. Анионы – простые и комплексные. Растворимость в
воде. Взрывчатые свойства. Распределение заряда на атомах азота. Ковалентные
производные.
2.
Диазотирование первичных
ароматических аминов. Механизм диазотирования (упрощённая схема с использованием
Н+ и NO+). Сколько молей кислоты требуется? (Формально – 2,
реально – больше.) Побочное образование триазенов и побочное азосочетание.
3.
Агенты диазотирования в
порядке уменьшения их реакционной способности.
NO+>>H2NO2+>NOBr>NOCl>N2O3>HNO2.
4.
Нитрозирование втор. и трет. аминов. Реакция алифатических аминов с HNO2.
5. Приемы
диазотирования: а) классический, б) для низкоосновных аминов, в) обратный
порядок смешения, г) в неводной среде - использование i-AmONO. Особенности диазотирования фенилендиаминов. Контроль
окончания реакции.
6. Поведение солей диазония в щелочной среде. Диазогидрат, син- и анти-диазотаты.
Амбидентность диазотатов.
|
7. Реакции
диазосоединений с выделением азота .
1) Термическое разложение
арилдиазония протекает через высокореакционноспособные арил-катионы.
Механизм замещения в этом случае аналогичен SN1 в алифатической химии. По этому механизму идет
реакция Шимана и образование фенолов и их простых эфиров.
2)
Нуклеофилы – восстановители. Механизм – перенос электрона, и образование
арил-радикала. По этому механизму протекает реакция с иодид-ионом, замещение
диазогруппы на водород.
3)
Реакции в присутствии порошка меди или солей меди(I). Также имеют радикальную природу, роль
восстановителя играет медь. Нуклеофил переносится на арил-радикал в координационной
сфере комплексов меди. Таких реакций большинство в химии солей диазония.
Реакция Зандмейера и ее аналоги.
4) Реакция Несмеянова.
5) Диарилиодониевые и бромониевые соли.
|
8. Реакции диазосоединений без
выделения азота. Восстановление.
Азосочетание, требования к азо- и диазокомпонентам. Примеры
азокрасителей (метилоранж).
|
9. Реакции
Гомберга-Бахмана и Мейервейна
Современная альтернатива – реакции кросс-сочетания, катализируемые
комплексами переходных металлов и реакция Хека. На 5++: кросс-сочетание с
солями диазония и диарилиодониевыми солями.
10. ДИАЗОМЕТАН. Получение, строение, реакции с кислотами, фенолами,
спиртами (различие в условиях), с кетонами и альдегидами.
ФЕНОЛЫ И ХИНОНЫ.
Большинство важнейших методов
синтеза фенолов известны из материалов предыдущих лекций:
1)
синтез через Na-соли сульфокислот;
2)
гидролиз арилхлоридов;
3)
через соли диазония;
4)
кумольный метод.
5)
гидроксилирование
активированных аренов по Фентону.
СВОЙСТВА ФЕНОЛОВ.
1) Кислотность; 2) синтез
сложных эфиров; 3) электрофильное замещение (см. тему "Электрофильное замещение
в аренах");
4) Реакции электрофильного замещения, не
рассмотренные ранее: карбоксилирование по Кольбе, формилирование по
Реймеру-Тиману, нитрозирование; 5) таутомерия, примеры; 6) Синтез простых
эфиров; 6а) синтез аллиловых эфиров; 7) перегруппировка Кляйзена;
8)
окисление фенолов, ароксильные радикалы; реакция
Бухерера;
10) превращение PhOHв PhNR2.
ХИНОНЫ.
1. Строение хинонов. 2. Получение хинонов. Окисление
гидрохинона, семихинон, хингидрон. 3. Хлоранил, 2,3-дихлор-5,6-дициано-1,4-хинон
(DDQ). 4. Свойства хинонов: а)
окислительно-восстановительные реакции, 1,2-
и 1,4-присоединение, реакция Дильса-Альдера.
ВАЖНЕЙШИЕ
ПРИРОДНЫЕ ЕНОЛЫ, ФЕНОЛЫ И ХИНОНЫ.
ВИТАМИН С (1): Аскорбиновая кислота. Восстановитель. Окрашивание с FeCl3. В природе синтезируется всеми
хлорофиллсодержащими растениями, пресмыкающимися и земноводными, многими
млекопитающими. Человек, обезьяны, морские свинки в ходе эволюции утратили
способность ее синтезировать.
Важнейшие функции – построение
межклеточного вещества, регенерация и заживление тканей, целостность
кровеносных сосудов, устойчивость к инфекции и стрессу. СИНТЕЗ КОЛЛАГЕНА
(гидроксилирование аминокислот). (Коллаген – это наше все: кожа, кости, ногти,
волосы.) Синтез норадреналина. Недостаток витамина С – ЦИНГА. Содержание
витамина С: черная смородина 200 мг/100 г, красный перец, петрушка –
150-200, цитрусовые 40-60, капуста –
50. Потребность: 50-100 мг/день.
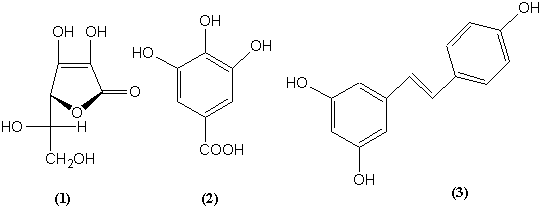
ТАННИН,
это гликозид галловой кислоты (2). Содержится
в чае, обладает дубящими свойствами
РЕЗВЕРАТРОЛ(3) – содержится в КРАСНОМ
ВИНЕ (французском). Снижает вероятность сердечно-сосудистых заболеваний.
Ингибирует образование пептида ЭНДОТЕЛИНА-1 – ключевого фактора в развитии
атеросклероза. Способствует продвижению французского вина на рынок. Более 300
публикаций за последние 10 лет.
ГВОЗДИЧНОЕ МАСЛО: эвгенол (4).
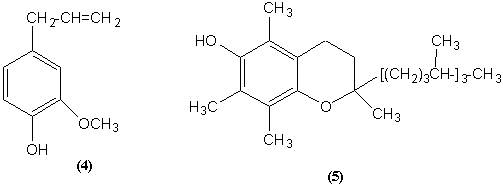
ВИТАМИН Е (5) (токоферол –
"потомство несу"). Антиоксидант. (Сам образует неактивные свободные радикалы). Регулирует обмен селена в глутатионпероксидазе
– ферменте, который защищает мембраны от пероксидов. При недостатке –
бесплодие, мышечная дистрофия, снижение потенции, возрастает окисляемость липидов
и ненасыщенных жирных кислот. Содержится в растительных маслах, салате,
капусте, желтке, злаках, овсянке (геркулес, мюсли). Потребность – 5 мг/день.
Авитаминоз бывает редко.
ВИТАМИНЫ ГРУППЫ К (6). Регуляция свертываемости крови и минерализации костной ткани
(карбоксилирование остатка глутаминовой кислоты в положение 4 (в составе
белков!)) - результат: связывание
кальция, рост костей. Синтезируется в
кишечнике. Потребность – 1 мг/сут. Геморрагические заболевания. Антивитамины К.
Дикумарин. Снижение свертываемости крови при тромбозах.
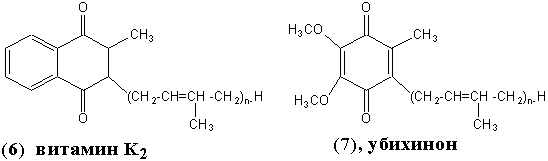
УБИХИНОН ("вездесущий хинон"), он же – коэнзим Q (7). Перенос электронов. Тканевое дыхание. Синтез
АТФ. Синтезируется в организме.
ХРОМОН (8) и ФЛАВОН (9) – полухиноны, полуэфиры фенолов.
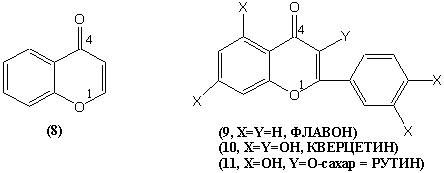
КВЕРЦЕТИН (10). РУТИН – витамин Р (11) (это кверцетин + сахар).
Витамин
проницаемости. При недостатке кровотечение, утомляемость, боли в конечностях. Связь витаминов С и Р (аскорутин).
АНТОЦИАНИНЫ (от греч.: окраска цветов).
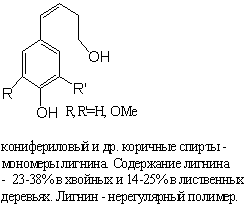
ЧТО ТАКОЕ ЛИГНИН? Из чего состоит древесина?
почему она твердая и водонепроницаемая?
"АЛИЦИКЛЫ", 2 лекции.
1.
Формальная классификация циклов (гетероциклы и карбоциклы, и те и другие могут быть ароматическими
и неароматическими. Алициклами называются неароматические карбоциклы.
2.
Распространение в
природе (нефть, терпены, стероиды, витамины, простагландины, хризантемовая к-та
и пиретроиды, др.).
3.
Синтез – конец XIX в. Перкин мл. – из натрмалонового эфира. (см. п.13).
Густавсон:
Br-CH2CH2CH2-Br + Zn (EtOH, 80oC). Это 1,3-элиминирование.
4.
БАЙЕР (1885 г.). Теория
напряжения. Это даже не теория, а дискуссионная статья: По Байеру – все циклы плоские. Отклонение от угла 109о28’ –
напряжение. Теория жила-жила лет 50, потом умерла, а термин – остался.
Первые синтезы макро- и средних циклов (Ружичка).
5.
ТИПЫ НАПРЯЖЕНИЯ В ЦИКЛАХ: 1) УГЛОВОЕ (только малые
циклы), 2) ТОРСИОННОЕ (заслоненное), ТРАНСАННУЛЯРНОЕ (в средних циклах).
|
|

|

|

|

|

|

|
|
Напр.
по Байеру
|
49o28’/2
=24o44’
|
9o44’
|
0o44’
|
5o16’
|
9о33’
|
12o46’
|
|
Напр. по DHof ккал/м
(тепл. образ.)
|
27,2
|
26,8
|
6,6
|
0
|
6
|
10
|
|
Напр. по DHof ккал/м: С9 (12,5 ккал/м), С10 (13
ккал/м), С11 (11 ккал/м), С12 (4 ккал/м), С14
(2 ккал/м).
|
|
Теплота сгорания на CH2 группу, ккал/м
|
МАЛЫЕ ЦИКЛЫ
166.6 (С3), 164,0 (С4)
|
ОБЫЧНЫЕ
158,7 (С5), 157,4 (С6)
|
СРЕДНИЕ ДО С12 (C13)
МАКРОЦИКЛЫ > C13
|
6. ЦИКЛОПРОПАН. Строение (С-С 0,151 нМ, ÐНСН = 114о), гибридизация (по расчётамдляС-Н она ближе к sp2, для C-C– к sp5), банановые
связи, угол 102о сходство с алкенами, ТОРСИОННОЕ напряжение – 1 ккал/м
на С-Н, т.е. 6 ккал/м из 27.2 (табл.). Кислотность CH – рКа как у этилена =36-37, возможноесопряжение
циклопропанового фрагмента с р-орбиталями
соседних фрагментов (стабильность циклопропилметильного карбокатиона).
ОСОБЕННОСТИ
ХИМИЧЕСКИХ СВОЙСТВ. 1. Гидрирование в С3Н8
(Н2/Pt, 50оС)/
2. с влажным HBr – раскрытие
цикла метилциклопропана по Марковникову, 1,5-присоединение к винилциклопропану
3. Радикальное галогенирование. 4. Устойчивость
к некоторым окислителям (нейтральный раствор KMnO4, озон).
В фенилциклопропане озон окисляет Phкольцо cобразованием
циклопропанкарбоновой кислоты.
7.
ЦИКЛОБУТАН. Строение (С-С 0,155 нМ, ÐНСН = 107о), КОНФОРМАЦИЯ – складчатая, отклонение от плоскости равно 25о.
ТОРСИОННОЕ напряжение.
Почти нет ОСОБЕННОСТЕЙ ХИМИЧЕСКИХ СВОЙСТВ:Гидрирование в С4Н10 (Н2/Pt, 180оС).
Особенности строения оксетанов: ТОРСИОННОЕ напряжение – 4 ккал/м вместо 8.
8.
ЦИКЛОПЕНТАН. Углового напряжения почти нет. В плоском – 10 пар заслоненных С-Н
связей (это могло бы дать торсионное напряжение 10 ккал/м, но циклопентан – не
плоский). Конформации: открытый КОНВЕРТ
– полукресло – открытый КОНВЕРТ. ПСЕВДОВРАЩЕНИЕ – компромисс между угловым и
торсионным напряжением.
9. ЦИКЛОГЕКСАН
– КРЕСЛО. Углового и торсионного напряжения нет. Аксиальные и экваториальные
атомы. Все С-Н связи соседних атомов
углерода находятся в в заторможенном положении. Переход между двумя возможными кресловидными конформациями через твист-форму, и т.д. 105 раз в сек.
ЯМР спектр циклогексана.Быстрые
и медленные обменные процессы в ЯМР.
МОНОЗАМЕЩЕННЫЕ
ЦИКЛОГЕКСАНЫ. Конформеры. Аксиальные и гош-бутановые взаимодействия.
Свободные
конформационные энергии заместителей. – DGо, ккал/м:
H(0), Me(1.74, это ~95%
е-Ме конформера в равновесии), i-Pr(2.1), t-Bu (5.5), Hal (0.2-0.5) Ph(3.1).
Трет-бутильная группа как якорь, закрепляющий конформацию,
в которой она сама занимает экваториальное положение. В трет-бутилциклогексане при комнатной температуре более 99,99% экваториального конформера.
Аномерный
эффект. Открыт на моносахаридах и
более подробно будет рассмотрен там.
10.
ДИЗАМЕЩЕННЫЕ ЦИКЛОГЕКСАНЫ. Цис- транс- изомеры, энантиомеры для 1,2-. 1,3-.
1,4-дизамещённых циклогексанов.
11. ВЛИЯНИЕ
КОНФОРМАЦИОННОГО СОСТОЯНИЯ на реакционную способность. Вспомнить элиминирование
в ментил- и изоментилхлориде (1 сем). Правило Бредта.
12. Понятие о
конформациях средних циклов (кресла-ванны, короны и т.п.) Трансаннулярное
напряжение. Понятие о трансаннулярных реакциях.
13. Методы
синтеза малых циклов.
|
ЦИКЛОПРОПАН.
а) из 1,3-дибромпропана по Густавсону.
б) 1,3-элиминирование из Cl(CH2)3CN,
в) малоновый синтез, превращение
циклопропанкарбоновой кислоты в циклопропилбромид по Бородину-Хунсдикеру.
г) присоединение дихлоркарбена к C=Cсвязи,
превращение ССl2 в СН2,
например с Li/t-BuOH).
д) Диазометан + C=C.
е) По Кижнеру из окиси мезитила и N2H4.
ж) Синтез трехчленных циклов по Кулинковичу.
|
ЦИКЛОБУТАН.
а)
Натрмалоновый эфир с 1,3-дибромпропаном.
б)
Ацилоиновая конденсация с Me3SiCl.
в)
Вспомнить про [2+2] циклоприсоединение. Когда запрещено термическое и
разрешено фотохимическое. Когда все же
бывает термическое (наличие сильных EWG у С=С).
|
14. СИНТЕЗ
ОБЫЧНЫХ И СРЕДНИХ ЦИКЛОВ.
Через малоновый эфир.
Пиролиз Ca, Ba, Mn, Th солей a,w –дикарбоновых кислот.
Конденсация
Дикмана.
Через a,w –динитрилы.
Ацилоиновая кондесация.
Метатезис алкенов.
Циклотри- и тетрамеризация на металлокомплексных
катализаторах.
Реакция Демьянова.
15.
Особенности строения циклоалкенов.
16. Синтез
циклоалкинов.
17. Бициклы.
Спираны. Адамантан.
18. Экзотика.
Тетраэдран, кубан, ангуланы, пропеллан.
ГЕТЕРОЦИКЛИЧЕСКИЕ СОЕДИНЕНИЯ.
1.
Пятичленные гетероциклы с одним гетероатомом.
Пиррол,
фуран, тиофен, ароматичность, их производные в природе (порфирин, гем,
хлорофилл, витамин В12, аскорбиновая кислота, биотин).
2. Методы синтеза пятичленных
гетероциклов с одним гетероатомом. Метод
Пааля-Кнорра. Синтез пиррола по Кнорру и
фурана по Фейсту-Бенáри. Превращения
фурана в другие пятичленные гетероциклы по Юрьеву. Получение фурфурола из растительных отходов, содержащих пятиуглеродные
углеводы (пентозаны).
3.
Физические и химические свойства пятичленных гетероциклов.
Данные спектров ЯМР 1Н и 13С,
δ м.д. (для бензола δН 7.27 и
δС 129 м.д.)
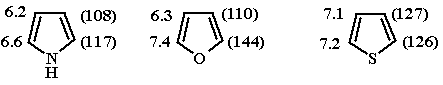
Дипольные моменты
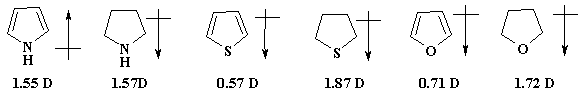
3.1
Электрофильное замещение в пирроле, фуране и тиофене.
По реакционной способности по отношению к электрофилам
пиррол напоминает активированные ароматические субстраты (фенол или
ароматические амины), Пиррол более реакционноспособен, чем фуран (фактор
скорости более 105), тиофен значительно менее реакционноспособен,
чем фуран (также приблизительно в 105 раз), но более
реакционноспособен, чем бензол (фактор скорости 103-105).
Все пятичленные гетероциклы склонны к полимеризации и осмолению в присутствии
сильных протонных кислот и высокореакционноспособных кислот Льюиса. Особенно
высокой ацидофобностью отличается пиррол. ДЛЯ ЭЛЕКТРОФИЛЬНОГО ЗАМЕЩЕНИЯ В ПЯТИЧЛЕННЫХ
ГЕТЕРОЦИКЛАХ, ОСОБЕННО ПИРРОЛЕ, НЕЛЬЗЯ БРАТЬ СИЛЬНЫЕ МИНЕРАЛЬНЫЕ КИСЛОТЫ, AlCl3, А ТАКЖЕ
СИЛЬНЫЕ ОКИСЛИТЕЛИ! Хотя это правило не абсолютно, и тиофен до некоторой
степени устойчив к действию кислот, проще и надежнее совсем избегать таких
реакций для всех донорных гетероциклов. Примеры реакций электрофильного
замещения в пирроле, фуране и тиофене.
3.2. Основность и кислотность пиррола, алкилирование Li, Na, К и Mg производных пиррола.
3.3.
Конденсация пиррола с альдегидами (формилирование, образование порфиринов).
3.4.
Особенности химических свойств фуранов (реакция с бромом, реакция Дильса-Альдера.
3.5.
Особенности химических свойств тиофена. Десульфуризация.
3.6. Реакции
С-металлированных пятичленных гетероциклов.
4. Конденсированные пятичленные
гетероциклы с одним гетероатомом.
4.1. Индолы в природе
(триптофан, скатол, серотонин, гетероауксин. Индиго.)
4.2 Синтез
индолов по Фишеру. Механизм.
4.3 Сравнение
свойств индола и пиррола. Аналогично
пирролу индол ацидофобен и очень
чувствителен к окислителям. Существенным отличием от пиррола является
ориентация электрофильного замещения в положение 3.
5. Пятичленные гетероциклы с двумя гетероатомами.Имидазол, амфотерность, таутомерия, использование при
ацилировании. Сравнение с амидинами. Имидазол
– донор и акцептор водородных связей. Это важно для химии ферментов, например
химотрипсина. Именно гистидиновый фрагмент химотрипсина переносит протон и
обеспечивает гидролиз пептидной связи.
6. Пиридин, ароматичность, основность (рКа 5,23; основность сравнима с анилином (рКа = 4,8),
но немного больше). рКа производных пиридина: 2-амино-Ру= 6,9, 3-амино-Ру = 6,0. 4-амино-Ру = 9,2. Это довольно сильное
основание. 4-нитро-Ру = 1,6;
2-циано-Ру= -0,26).
Производные
пиридина в природе (витамины, никотин, NADP).
6.1. Данные спектров ЯМР 1Н
(13С), δ, м.д.
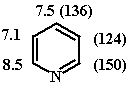
6.2. Методы синтеза пиридинов (из 1,5-дикетонов,
трёхкомпонентный синтез Ганча).
6.3. Химические свойства пиридина. Алкилирование, ацилирование, DMAP, комплексы пиридина с кислотами Льюиса. (cSO3, BH3, NO2+BF4-, FOTf). Мягкие электрофильные реагенты для сульфирования,
восстановления, нитрования и фторирования соответственно.
6.4. Реакции электрофильного замещения для пиридина. Особенности реакций и примеры условий
электрофильного замещения в пиридине.
6.5. N-окись
пиридина, получение и её использование в синтезе. Введение нитрогруппы в 4- положение кольца.
6.6. Нуклеофильное замещение в 2-, 3-, и
4-хлорпиридинах. Факторы парциальных
скоростей по сравнению с хлорбензолом.
Аналогичная
тенденция наблюдается и для 2-, 3- и 4- галогенхинолинов.
6.7. Нуклеофильное замещение гидрид-иона:
реакция пиридина с алкил- или
ариллитием;
реакция пиридина с амидом натрия
(реакция Чичибабина). Так как элиминирование
свободного гидрид-иона невозможно по энергетическим причинам, в реакции
Чичибабина промежуточный сигма-комплекс ароматизуется за счет взаимодействия с
продуктом реакции с образованием натриевой соли продукта и молекулярного
водорода.
В
других реакциях гидрид обычно удаляют с помощью окисления. Так, соли
пиридиния могут подвергаться гидроксилированию, приводящему к образованию
1-алкилпиридонов-2. Процесс идет аналогично аминированию, но в присутствии окислителя,
например, K3[Fe(CN)6].
6.8. Литиопроизводные пиридина. Получение, реакции.
6.9. Пиридиновое ядро как сильный мезомерный акцептор. Устойчивость
карбанионов, сопряженных с пиридиновым ядром в 2- или 4-положениях. Особенности
химических свойств метилпиридинов и винилпиридинов.
7. Конденсированные шестичленные гетероциклы с одним
гетероатомом.
7.1. Хинолин. Хинин.
Спектры ЯМР 1Н (13С) хинолина,
δ, м.д.
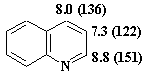
7.1. Методы получения хинолинов. Синтезы Скраупа и
Дёбнера-Мил-лера. Понятие о механизме
этих реакций. Синтез 2- и 4-метилхинолинов.
7.2. Изохинолины, синтез
по Бишлеру-Напиральскому.
7.3. Химические свойства хинолинов и изохинолинов. Сравнение с пиридином, отличия свойств пиридина и
хинолина.
Поведение гетероциклических
соединений в присутствии окислителей и восстановителей, предназначенных для модификации
боковых цепей.
Восстановители:
Пиррол – практически неограниченно
устойчив к действию восстановителей, а также оснований и нуклеофилов (например,
выдерживает гидриды, боран, Na в спирте без
затрагивания кольца, даже при длительном нагревании).
Тиофен – также как и пиррол, устойчив
к действию восстановителей, а также оснований, нуклеофилов за исключением восстановителей на основе переходных
металлов. Любые соединения никеля (никель Ренея, борид никеля) вызывают
десульфуризацию и гидрирование скелета. Катализаторы на основе палладия и
платины обычно отравляются тиофенами и не работают.
Фуран – также как пиррол, но
очень легко гидрируется.
Индол – полностью аналогичен
пирролу.
Кольцо
пиридина восстанавливается легче, чем кольцо бензола. Для боковых цепей можно
использовать NaBH4, нежелательно (часто даже нельзя) использовать LiAlH4.
Для хинолина закономерности
практически те же, что для пиридина, нельзя использовать LiAlH4.
В кватернизованной форме (N-алкилпиридиний, хинолиний) очень чувствительны к
восстановителям (восстановление кольца), основаниям, нуклеофилам (раскрытие
кольца).
Окислители.
Использование окислителей для
соединений пиррола, индола и, в меньшей степени фурана, приводит, как правило,
к разрушению кольца. Присутствие электроноакцепторных заместителей повышает
устойчивость к окислителям, однако более подробная информация об этом выходит
за пределы программы 3 курса.
Тиофен ведет себя подобно
бензолу – обычные окислители не разрушают кольцо. Но категорически исключено
использование перекисных окислителей в любой форме – происходит окисление серы
до сульфоксида и сульфона с потерей ароматичности и немедленной димеризацией.
Пиридин достаточно устойчив к
большинству окислителей в мягких условиях. Отношение пиридина к нагреванию с KMnO4 (рН 7)
до 100oС в запаянной ампуле такое
же, как и для бензола: кольцо окисляется. В кислой среде в протонированной
форме пиридин еще более устойчив к окислителям, можно использовать стандартный
набор реагентов. Надкислоты окисляют пиридин в N-оксид – см. выше.
Окисление одного из колец хинолина с KMnO4 приводит к пиридин-2,3-дикарбоновой кислоте.
8. Шестичленные гетероциклы с несколькими атомами
азота
8.1.
Пиримидин. Производные пиримидина как
компоненты нуклеиновых кислот и лекарственных препаратов (урацил, тимин,
цитозин, барбитуровая кислота). Противовирусные
и противоопухолевые препараты – пр-ные пиримидина (5-фторурацил, азидотимидин,
алкилметоксипиразины – компоненты запаха пищи, фруктов, овощей, перца, гороха,
жареного мяса. Так называемая реакция Мейяра (Maillard)
(факультативно).
8.2. Понятие о химических свойствах
производных пиримидина.
Пиримидин
можно бромировать в положение 5. Урацил (см. ниже) тоже можно бромировать и
нитровать в положение 5.
Легкие реакции SN2Arв
хлорпиримидинах (аналогия с
пиридином!): по положению 4 идёт быстрее,
чем по 2.
Замещение 2-С1 под действием КNH2 в NH3 ж. Механизм
не ариновый, а ANRORC (на 5+++).
10.
Биядерные гетероциклы с несколькими атомами азота.
Пурины (аденин, гуанин).
Наиболее
известные пурины (кофеин, мочевая кислота, ацикловир). Изостеры пуринов
(аллопуринол, силденафил ( виагра™)).
Дополнительная
литература по теме "Гетероциклы"
1.
Т.Джилкрист "Химия гетероциклических соединений" (Пер. с англ. - М.: Мир, 1996)
2.
Дж. Джоуль, К.Миллс "Химия гетероциклических соединений" (Пер. с англ. - М.:
Мир, 2004).
АМИНОКИСЛОТЫ.
1.Аминокислоты
(АК) в природе. (≈ 20 аминокислот присутствуют в белках, это кодируемые АК, >200 АК встречаются в
природе.)
2.
α-, β-, γ-аминокислоты.
S-конфигурация
природных L-аминокислот.
3.
Амфотерность, изоэлектрическая точка (рН
обычно 5,0-6,5). Оснόвные (7,6-10,8), кислые (3,0-3,2) аминокислоты. Подтверждение
цвиттер-ионного строения. Электрофорез.
4.
Химические свойства АК – свойства COOHи NH2 групп.
Хелаты. Бетаины. Поведение при нагревании
(ср. с оксикислотами). Образование
азлактонов из N-ацетилглицина и гидантоинов из мочевины и АК – на 5++. Синтез сложных
эфиров и N-ацилирование
– путь к пептидному синтезу (см.
лекцию про белок).
5.
Химическое и биохимическое
дезаминирование, (механизмы не учить!), принцип ферментативного переаминирования
с витамином В6 (было в теме
"Карбонильные соединения" и в курсе биохимии).
6.
Декарбоксилирование – термическое и
ферментативное.
7. Важнейшие способы синтеза
аминокислот:
1)
из галогенкарбоновых кислот – два примитивных способа, включая фталимидный.
(Оба уже известны!)
2)
синтез Штрекера;
3)
алкилирование анионов СН-кислот – PhCH=N–CH2COOR и N-ацетиламиномалонового
эфира.
4)
Энантиоселективный синтез АК путем:
а)
микробиологического (ферментативного) разделения и
б)
энантиоселективное гидрирование с использованием хиральных катализаторов.
5)
β-аминокислоты. Синтез по Михаэлю.
|
Гидрофобные аминокислоты
|
Немного о биохимической роли (для
общего развития)
|
|
АЛАНИН
Ala (A)
|
|
Вынос аммиака из тканей в
печень. Трансаминирование, превращение в пировиноградную к-ту. Синтез
пуринов, пиримидинов и гема.
|
|
ВАЛИН*
Val (V)
|
|
Если в результате мутации валин встаёт на место глутаминовой к-ты в
гемоглобине – бывает наследственное заболевание –серповидно-клеточная анемия.
Серьёзная наследственная болезнь, распространенная в Африке, но при этом
придающая устойчивость к малярии.
|
|
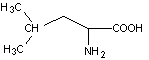
ЛЕЙЦИН*
Leu (L)
|
|
|
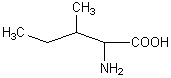
ИЗОЛЕЙЦИН*
ILe (I)
|
|
|
ПРОЛИН
Pro (P)
|
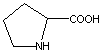
|
Изгибы в молекулах белков. Отсутствие вращения там,
где есть пролин.
|
|
ФЕНИЛАЛАНИН*
Phe (F)
|
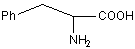
|
Если не
превращается в тирозин – будет наследственное заболевание фенилпировино-градная
олигофрения.
|
|
ТРИПТОФАН*
Trp (W)
|
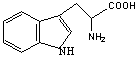
|
Синтез НАДФ, серотонина. Распад
в кишечнике до скатола и индола.
|
|
|
Гидрофильные аминокислоты
|
|
|
ГЛИЦИН Gly (G)
|
H2N-CH2-COOH
|
Участвует в огромном кол-ве
биохимических синтезов в организме.
|
|
СЕРИН Ser (S)
|
HO-CH2-CH(NH2)-COOH
|
Участвуют (в составе белков) в
процессах ацилирования и фосфорилирования.
|
|
ТРЕОНИН* Thr (T)
|
CH3-CH(OH)- CH(NH2)-COOH
|
|
ТИРОЗИН Tyr (Y)
|
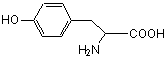
|
Синтез тиреоидных гормонов,
адреналина и норадреналина
|
|
|
"Кислые" аминокислоты
|
|
|
АСПАРАГИНОВАЯ КИСЛОТАAsp (D)
|
HOOC-CH2-CH(NH2)-COOH
|
Донор аминогруппы при синтезах.
|
|
ГЛУТАМИНОВАЯ КИСЛОТАGlu (E)
|
HOOC-C4H2-CH2-CH(NH2)-COOH
|
Образует ГАМК (γ-аминомасляную
кислоту (аминалон) – успокоительное
средство. Gluвыносит NH3 из мозга, превращаясь при этом в глутамин
(Gln). 4- карбоксиглутаминоваяк-тавбелкахсвязываетСа.
|
|
|
"А М И Д Ы" кислых аминокислот
|
|
|
АСПАРАГИН Asn (N)
|
H2N-CO-CH2-CH(NH2)-COOH
|
|
|
ГЛУТАМИН Gln (Q)
|
H2N-CO-CH2-CH2-CH(NH2)-COOH
|
Донораминогрупп в синтезах
|
|
|
Серосодержащие
|
|
|
ЦИСТЕИН Cys (C)
|
HS-CH2-CH(NH2)-COOH
|
Образование S-S связей (трет, структура белков, регуляция активности
ферментов)
|
|
ЦИСТИН
|
Cys-S-S-Cys
|
|
МЕТИОНИН*Met
|
MeSCH2CH2-CH(NH2)COOH
|
Донор метильных групп
|
|
|
"Оснóвные" аминокислоты
|
|
|
ЛИЗИН*Lys (K)
|
H2N-(CH2)4-CH(NH2)-COOH
|
Образует сшивки в коллагене и
эластине делает их эластичными.
|
|
АРГИНИНArg (R)
Содержит
фрагмент гуанидина
|
H2N-C(=NH)-NH-(СН2)3-СH(NH2)-COOH
|
Участвует в выведении аммиака
из организма
|
|
ГИСТИДИНHis (H)
Остаток
имидазола
|
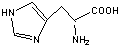
|
Синтез гистамина. Аллергия.
|
* - незаменимые аминокислоты. Из большинства аминокислот легко синтезируются глюкоза
и жиры. Нарушения обмена аминокислот у детей приводит к умственной
неполноценности.
ЗАЩИТНЫЕ ГРУППЫ, ИСПОЛЬЗУЕМЫЕ В ПЕПТИДНОМ СИНТЕЗЕ.
NH2-защитные
группы –
RC(O)- = (HC(O)- ) CF3C(O)- фталильная
ROC(O)- = PhСН2OC(O)- и замещённые бензилы, t-BuOC(O)- и др. трет-группы,
Флуоренилметилоксикарбонильная
группа,
Ts-группа
СООН -защитные группы – эфиры – PhCH2O- и замещенные
бензилы,
t-BuO- и
флуоренилметиловые эфиры.
Отдельное рассмотрение защитных групп для других ФГ
аминокислот не предусмотрено.
Методы
создания пептидной связи.
1. Хлорангидридный (через X-NH-CH(R)-C(O)Cl). Способ
устарел.
2..Азидный
(по Курциусу, через X-NH-CH(R)-C(O)Y → C(O)N3 как мягкий ацилирующий реагент.
3.Ангидридный
– напр. через смешанный ангидрид с угольной кислотой.
4.Активированные
эфиры (например С(О)-ОС6F5 и т.п.)
5.
Карбодиимидный – кислота + ДЦК + амин
6.
Синтез на твёрдом
носителе (напр. на смоле Меррифилда).
Биологическая
роль пептидов. Несколько примеров.
1.
Энкефалины и эндорфины – опиоидные пептиды.
например
Tyr-Gly-Gly-Phe-Met и
Tyr-Gly-Gly-Phe-Leu измозгасвиньи. Известно несколько сотен аналогов.
2.
Окситоцин и вазопрессин Cys-Tyr-Ile-Gln-Asn-Cys-Pro-Leu-Gly-NH2
│________________│
DuVigneaud, Ноб.пр.
1955 Cys-Tyr-Ile-Gln-Asn-Cys-Pro-Arg-Gly-NH2
│________________│
3.
Инсулин контролирует усвоение глюкозы клеткой. Избыток глюкозы в крови (диабет) – приводит
к гликозилированию всего подряд (в основном белков).
4.
Превращения пептидов: ангиотензиноген →
ангиотензин I →
ангиотензин II. Один из
основных механизмов регуляции артериального давления (АД), место приложения
многих лекарств (блокаторы АПФ – ангиотензин-превращающего фермента.
Катализатор 1 стадии – фермент ренин (выделен из почек).
5.
Пептидные токсины. Действуют при болезнях - ботулизм,
столбняк, дифтерия, холера. Яды змей, скорпионов, пчёл, токсины грибов
(фаллоидин, амантин), морских беспозвоночных (Conusgeographus – 13 АК, два -S-S-мостика).
Многие устойчивы при кипячении в кислом растворе (до 30 мин).
6.
Пептидные антибиотики (грамицидин S).
7.
Аспартам Asp-Phe-OMe в 200 раз слаще сахара. Горькие и "вкусные" пептиды.
8.
Белки. Четыре уровня
организации нативной белковой молекулы. Белок – уникальный (наряду с
нуклеиновыми кислотами) тип макромолекулы, обладающей точно известной
структурой, упорядоченной вплоть до деталей стереохимии и конформации. Все
остальные известные макромолекулы, в том числе и природные (полисахариды,
лигнин и т.п.) имеют в большей или меньшей степени неупорядоченное строение – широкое
распределение молекулярных масс, свободное конформационное поведение.
Первичная структура – последовательность
аминокислот. Как кратко обозначается первичная структура?
Вторичная структура –
конформационно-регулярные элементы двух типов (α–спирали и β– слои) –
так упорядочивается только часть белковой макромолекулы.
Третичная структура –
уникальная упорядоченная стереохимическая конфигурация полной макромолекулы.
Понятие о "складывании" (folding)
полипептидной цепи в третичную структуру белка. Прионы.
Четвертичная структура –
соединение нескольких субъединиц в белках, состоящих из нескольких полипептидных
цепей. Дисульфидные мостики (обратимое превращение цистеин-цистин) как способ
закрепления третичной и четвертичной структуры.
УГЛЕВОДЫ.
1.
Что такое углеводы? Углеводы вокруг и внутри нас.
2.
Понятие о фотосинтезе производных D-глицериновой кислоты.Только для особо выдающихся студентов –
об образовании дифосфата глицериновой кислоты из D-рибулозы.
3.
Что такое D-ряд
углеводов. (Кратко об истории возникновения понятия о D- и L- рядах).
4.
Классификация углеводов: а) по количеству атомов С; б)
по наличию С=О или СНО групп; в) по количеству циклических фрагментов.
5.
Синтез углеводов из D-глицеринового
альдегида по методу Килиани-Фишера. Как Фишер установил формулу глюкозы?
6.
Вывод формул всех D-тетроз,
-пентоз, -гексоз из D-
глицеринового альдегида (открытые структуры). Для всех студентов – знать формулу
глюкозы (открытую и циклическую), маннозы (2-эпимер глюкозы), галактозы
(4-эпимер глюкозы), рибозы. Пиранозы и фуранозы.
7.
Уметь перейти от открытой формы к циклической по
Хеуорсу. Уметь нарисовать формулы α- и β–глюкозы (все заместители в
е- положении кроме аномерного) в конформации кресла.
8.
Что такое эпимеры, аномеры, мутаротация. Аномерный эффект.
9.
Химические свойства глюкозы как альдегидоспирта: а)
хелаты с ионами металлов, получение гликозидов, полных простых и сложных
эфиров, изопропилиденовая защита; б)
окисление СНО группы ионами металлов, бромной водой, НNО3. Расщепление по Волю. Реакция с аминами и получение озазонов. Важнейшие принципы и приёмы избирательного
алкилирования различных гидроксилов в глюкозе.
10.
D-фруктоза
как представитель кетоз. Открытая и циклическая формы. Реакция серебряного зеркала для фруктозы.
11.
Понятие о
дезоксисахарах, аминосахарах. Сюда же хитин и гепарин. Септулозы и октулозы в
авокадо. Реакция Мейяра (Maillard).
12.
ОЛИГОСАХАРИДЫ. Мальтоза, целлобиоза, лактоза,
сахароза. Восстанавливающие и невосстанавливающие сахара.
13.
Полисахариды –
крахмал (20% амилозы + 80% амилопектина),
иодкрахмальная проба, гликоген, целлюлоза, гидролиз крахмала в полости рта
(амилаза) и гидролиз целлюлозы, нитроклетчатка, вискозное волокно, производство бумаги, группы крови и отличие между ними.
ВАЖНЕЙШИЕ ПОЛИСАХАРИДЫ.
|
ПОЛИСАХАРИД
|
СОСТАВ и
строение
|
примечания
|
|
циклодекстрины
|
α-(6),
β-(7), γ-(8)
Состоит из
глюкозы,
1-4 связи.
|
Отличные
комплексообразователи, хелатообразователи
|
|
крахмал
|
α-глю-(1,4)-α-глю
20%
амилозы + 80% амилопектина
|
Амилоза = 200 глю, линейный полисахарид.
Амилопектин
= 1000 и более глю, разветвлён.
|
|
гликоген
|
"разветвлённый"
крахмал, участие 6-ОН
|
Запас
глюкозы в организме
|
|
инулин
|
Из остатков фруктозы
|
Содержится в топинамбуре
|
|
целлюлоза
|
β-глю-(1,4)-β-глю
|
Хлопок,
клетчатка растений, древесина
|
|
целлюлоза
|
Ксантогенат
по 6-положению
|
Получение вискозы – искусственного шелка, целлофана
(упаковочной плёнки)
|
|
ацетат
целлюлозы
|
Примерно
диацетат
|
ацетатное
волокно
|
|
нитрат
целлюлозы
|
Тринитроэфир
|
Бездымный
порох
|
|
Производство бумаги из древесины
|
Древесина =
целлюлоза + лигнин.
Обработать Са(НSO3)2
или Na2S + NaOH
|
Сульфатация древесины – удаление лигнина в воду –
получение целлюлозной массы.
|
|
хитин
|
Поли-α-2-дезокси-2-N-Ac-аминоглюкоза
(вместо 2-ОН – 2-NH-Ac)
|
Если удалить Ас от азота получится хитозан – модная
БАД
|
|
гиалуроновая кислота
|
– (2-AcNH-глюкоза –
глюкуроновая кислота)n –
|
Смазка в организме (напр. в суставах).
|
|
гепарин
|
Строение очень сложное – (2-HO3S-NH-глюкоза – глюкуроновая кислота)n –
|
Увеличивает время свёртываемости крови
|
|
Хондроитин сульфат
|
Гликопротеины (коллаген), протеогликаны, связь через
NН2 аспарагина или ОН серина
|
Есть везде в организме, особенно в соединительной
ткани, хрящах.
|
Примечание: Глюкуроновая
к-та: 6-СООН – 1- СНО
Глюконовая к-та: 6-СН2ОН –
1-СООН
Глюкаровая к-та: 6-СООН – 1- СООН
1.
Химия и биохимия нуклеиновых кислот.
Азотистые основания в РНК: У (урацил), Ц (цитозин) – производные пиримидина. А
(аденин), Г (гуанин) – производные пурина. В
ДНК вместо У (урацила) присутствует Т (тимин).
Нуклеозиды
(сахар + азотистое основание):
уридин, цитидин, тимидин, аденозин, гуанозин.
Нуклеотиды(фосфат
+ сахар + азотистое
основание).
Лактим-лактамная
таутомерия.
Первичная структура нуклеиновых кислот (соединение нуклеозидов через атомы
кислорода у С-3 и С-5 рибозы (дезоксирибозы) с помощью фосфатных мостиков.
РНК и ДНК.
а)
Главные основания и минорные основания (РНК). Только для тРНК список минорных
оснований приближается к 50. Смысл их существования – защита от гидролитических
ферментов. 1-2 примера минорных оснований.
в)
Правила Чаргаффа для ДНК. Важнейшее: А=Т. Г=Ц. Однако Г+Ц < А+Т для животных и растений.
•
Принципы строения
ДНК
•
1. Нерегулярность.
Существует
регулярный сахарофосфатный остов, к которому присоединены азотистые основания.
Их чередование нерегулярно.
•
2. Антипараллельность.
ДНК состоит
из двух полинуклеотидных цепей, ориентированных антипараллельно. 3`-конец одной
расположен напротив 5`-конца другой.
•
3. Комплементарность (дополнительность).
Каждому
азотистому основанию одной цепи соответствует строго определенное азотистое
основание другой цепи. Соответствие задается химией. Пурин и пиримидин в паре
образуют водородные связи. В паре A-Т две водородные связи, в паре Г-Ц – три,
поскольку в этих основаниях в ароматическом кольце имеется дополнительная
аминогруппа.
•
4. Наличие регулярной вторичной структуры.
Две
комплементарные, антипараллельно расположенные полинуклеотидные цепи образуют
правые спирали с общей осью.
Отличия между ДНК и РНК.
|
|
ДНК
|
РНК
|
|
Сахар
|
Дезоксирибоза
|
Рибоза
|
|
Азотистые основания
|
А, Т, Г, Ц
|
А, У, Г, Ц
|
|
Вид и количество цепей в молекуле
|
Линейные или кольцевые молекулы;
99.99% двойная спираль
|
Линейные молекулы;
одноцепочечная,
сложенная в третичную структуру за счет самокомплементарных участков
|
|
Важнейшие
виды РНК
|
Размер в нуклеотидах (E. coli)
|
|
мРНК - информационные (матричные) РНК 2-3%
|
75-3000
|
|
тPHK - транспортные РНК 15-16%
|
75-90
|
|
рРНК - рибосомные РНК 80-85%
|
120 – 1500 – 3100
|
|
Рибозимы (аналоги энзимов)
|
|
Функции
ДНК
1. ДНК является носителем генетической
информации.
Функция обеспечивается фактом существования генетического кода. Количество молекул ДНК: в клетке человека – 46
хромосом, в каждой одна молекула ДНК. Длина 1 молекулы ~ 8 (т.е.2х4) см. В упакованном виде – 5 нм (это
третичная структура ДНК, суперспирализация ДНК на белках-гистонах).
2. Воспроизведение и передача генетической
информации обеспечивается процессом репликации (ДНК → новая ДНК).
3. Реализация генетической информации в виде белков и любых других соединений,
образующихся с помощью белков-ферментов.
Эта функция обеспечивается процессами транскрипции (ДНК в РНК) и трансляции (РНК в белок).
Репарация –
восстановление повреждённого участка ДНК. Это происходит из-за того, что ДНК –
двухцепочечная молекула, есть комплементарный нуклеотид, который
"подсказывает", что нужно исправить.
Какие бывают ошибки и повреждения? а) Ошибки репликации (10-6),
б) депуринизация, потеря пурина, образование апуриновых сайтов (в каждой клетке
потеря 5000 пуриновых остатков в сутки!), в) дезаминирование (например цитозин
превратился в урацил).
Индуцируемые
повреждения. а) димеризация пиримидиновых колец под действием УФ по
С=С связям с образованием циклобутанового кольца (для удаления димеров
используются фотолиазы); б) химическое повреждение (алкилирование, ацилирование
и др.). Ремонт повреждений – ДНК-гликозилаза – апуринизация (или апиримидинизация)
алкилированного основания – далее введение "нормального" основания в пять
стадий.
Нарушение
процесса репарации – наследственные болезни (пигментная ксеродерма,
трихотиодистрофия и др.) Наследственных болезней около 2000.
Ингибиторы
транскрипции и трансляции – антибактериальные препараты.
Стрептомицин – ингибитор
синтеза белка у прокариотов.
Тетрациклины –
"связываются с 30S субъединицей
бактериальной рибосомы и блокируют присоединение аминоацил-тРНК в А-центр
рибосомы, тем самым, нарушая элонгацию (т.е. считывание мРНК и синтез
полипептидной цепи)".
Пенициллины
и цефалоспорины – β-лактамные антибиотики. β-Лактамное
кольцо ингибирует синтез клеточных стенок у грамотрицательных микроорганизмов.
Вирусы
– ингибиторы матричных синтезов в эукариотических клетках.
Токсины – часто
делают то же самое, что и вирусы. α–Аманитин
– токсин бледной поганки, LD50 0,1 мг на кг
массы тела. Ингибирование РНК-полимеразы. Результат – необратимые изменения в
печени и почках.
Рицин – очень
сильный белковый яд из клещевины. Это фермент N—гликозилаза,
который удаляет остаток аденина из 28S рРНК большой
субъединицы рибосомы, ингибирует синтез белка у эукариотов. Содержится в
касторовом масле.
Энтеротоксин
возбудителя дифтерии (белок с массой 60 кД) – ингибирование синтеза белков
в зеве и гортани.
Интерфероны
– белки с размером около 160 АК, секретируются некоторыми клетками
позвоночных в ответ на заражение вирусами. Количество интерферона – 10-9
– 10-12 г, т.е. одна молекула белка защищает одну клетку. Эти белки,
как белковые гормоны, стимулируют синтез ферментов, которые разрушают синтез
мРНК вирусов.
Наследственные болезни (моногенные) и
(не путать!) семейная предрасположенность к болезням (диабет, подагра,
атеросклероз, мочекаменная болезнь,
шизофрения – это мультифакторные болезни.)
Принципы анализа
нуклеотидной последовательности (факультативно).
ДНК
технологии в медицине.
А. Выделение ДНК. Б. Расщепление ДНК с
помощью рестриктаз. ДНК человека - 150х106 пар нуклеотидов. Их надо
разделить на 500 000 фрагментов по 300 пар в каждом. Далее гель-электрофорез.
Далее – блот-гибридизация по Саузерну с радиозондом или другие методики.
Секвенирование. Экзонуклеазы – отщепляют
последовательно один мононуклеотид. Это устаревшая методика.
ПЦР (PCR) – полимеразно-цепная реакция.
(Нобел. пр. 1993 г.: Кэрри Муллис)
Принцип: праймеры
(это фрагменты ДНК ~20 нуклеотидов – коммерчески доступные) +
ДНК-полимераза → наработка ДНК (амплификатор) → анализ ДНК
(секвенатор). Сейчас всё делается автоматически!
Метод секвенирования ДНК с использованием меченых дефектных нуклеотидов
(например, дидезоксинуклеотидов). Сейчас метки не радиоактивные, а
флуоресцентные. Анализ на СПИД и другие ИППП. Быстро, но дорого. Лучше не
болеть!
Успех ПЦР для диагностики и широкого распространения связан с тем, что
ферменты, участвующие в процессе, выделенные из термостойких бактерий горячих источников и сделанные генной
инженерией, выдерживают нагревание, при
котором происходит денатурация (диссоциация цепей ДНК) и подготовка их к
следующему циклу ПЦР.
ТЕРПЕНЫ, ТЕРПЕНОИДЫ И СТЕРОИДЫ.
Турпентин – летучее масло
из сосновой смолы.
Терпены
– группа ненасыщенных углеводородов состава (С5Н8)n, где n³ 2, широко распространенные в природе. Содержат
фрагменты изопентана, связанные, как правило, по типу “голова к хвосту” (это и есть Правило Ружички).
Монотерпены С10 (С5Н8)2 Сесквитерпены С15, (С5Н8)3 Дитерпены С20, (С5Н8)4 Тритерпены С30, (С5Н8)6 . Политерпены
(каучук).
Степень
гидрирования терпенов может быть разной, поэтому число атомов Н не обязательнодолжно
быть кратно 8. Не бывает С25 и
С35 терпенов.
Терпены
бывают ациклические и карбоциклические.
Терпеноиды
(изопреноиды) – это терпены (углеводороды) + функционально замещенные терпены.
Обширная группа природных соединений с регулярным строением скелета.
Изопреноиды можно подразделить на
1) терпены, в т.ч. функционально
замещенные,
2) стероиды
3) смоляные кислоты,
4) полиизопреноиды (каучук).
Важнейшие представители терпенов.
Некоторые особенности химии терпенов,
бициклических молекул и стероидов.
1) неклассические катионы; 2)
перегруппировки типа Вагнера-Мейервейна; 3) легкая окисляемость; 4)
диастереоселективный синтез; 5) влияние удаленных групп.
Формально
терпены – продукты полимеризации изопрена, но путь синтеза совершенно иной!
Почему же именно производные полиизопрена получили такое распространение в
природе? Это связано с особенностями их биосинтеза из ацетилкоэнзима А, т.е.
фактически из уксусной кислоты. (Блох, 40-60 гг. Оба атома углерода из С14Н3С14ООН
включаются в терпен.)
СХЕМА
СИНТЕЗА МЕВАЛОНОВОЙ КИСЛОТЫ – важнейшего промежуточного продукта в биосинтезе
терпенов и стероидов.
Конденсация
ацетилкоэнзима А в ацетоацетилкоэнзим А проходит по типу
сложноэфирной конденсации Кляйзена.
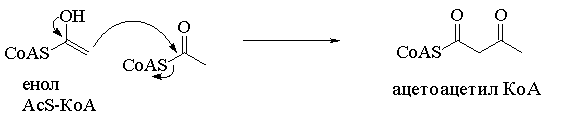
Далее
происходит реакция типа альдольной
конденсации:
Полученная молекула нехиральна, однако
она подвергается энантиоселективному ферментативному гидролизу только по одной
(левой) тиоэфирной группе. Дальнейшее восстановление другой тиоэфирной группы с
НАДФН(Н+) (справа) приводит к мевалоновой кислоте.
Синтез лимонена из геранилфосфата – важного промежуточного вещества как в
синтезе самых разнообразных терпенов, так и в синтезе холестерина. Ниже
приведено превращение лимонена в камфору под действием HCl, воды и окислителя (РР – остаток пирофосфата).
 Превращение мевалоновой кислоты в геранилфосфат происходит
путём 1) фосфорилирования 5-ОН, 2) повторного фосфорилирования 5-ОН и образование
пирофосфата, 3) фосфорилирования по 3-ОН. Всё это происходит под действием АТФ,
которая превращается в АДФ. Дальнейшие
превращения: Превращение мевалоновой кислоты в геранилфосфат происходит
путём 1) фосфорилирования 5-ОН, 2) повторного фосфорилирования 5-ОН и образование
пирофосфата, 3) фосфорилирования по 3-ОН. Всё это происходит под действием АТФ,
которая превращается в АДФ. Дальнейшие
превращения:
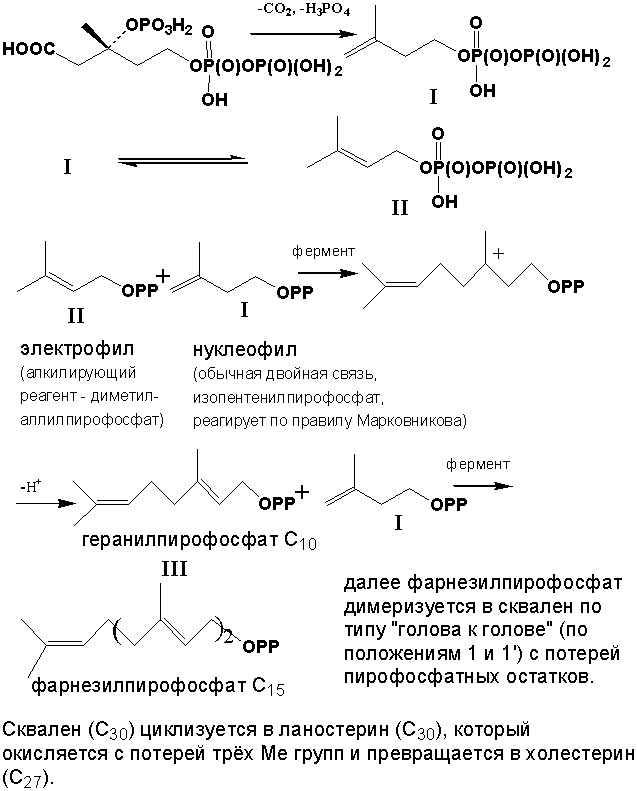
Важнейшие стероидные гормоны.
Образуются в организме из
холестерина. Холестерин не растворим в воде. Проникает в клетку и участвует в
биосинтезе через комплексы со стеринопереносящими белками.
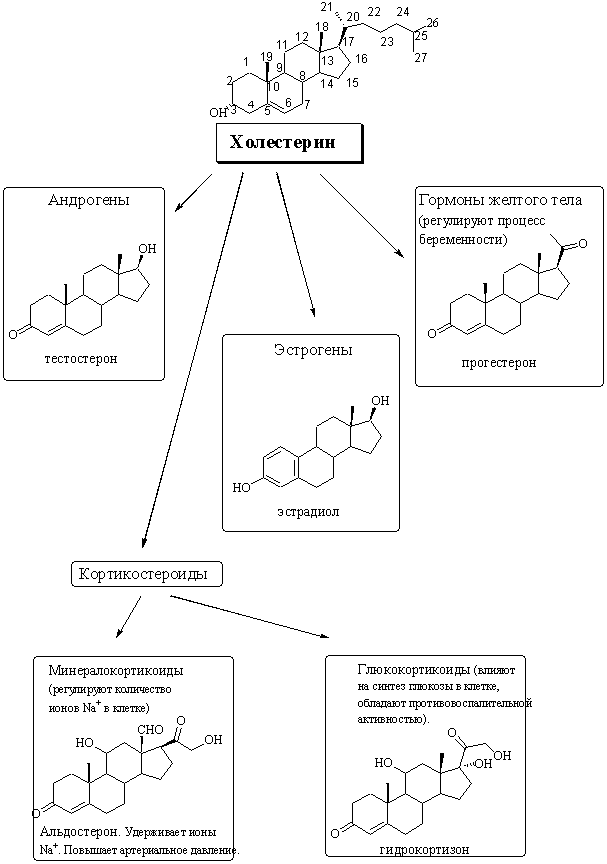
ЖЕЛЧНЫЕ
КИСЛОТЫ. Холевая кислота. Цис-сочленение колец А и В. Желчные
кислоты улучшают всасываемость липидов, понижают уровень холестерина, широко
используются для синтеза макроциклических структур.
СТЕРОИДЫ
– ЛЕКАРСТВЕННЫЕ СРЕДСТВА.
1.
Кардиотоники. Дигитоксин. Содержится в различных видах наперстянки (DigitalispurpureaL. или DigitalislanataEhrh.) Гликозиды
– это природные соединения, которые состоят из одного или нескольких остатков
глюкозы или другого сахара, чаще всего связанных через положения 1- или 4- с органической молекулой (АГЛИКОНом).
Вещества похожей структуры и действия встречаются в яде некоторых видов жаб.
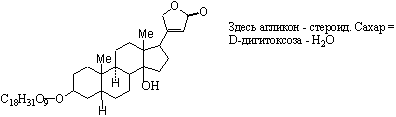
2. Диуретики.
Спиронолактон (верошпирон). Антагонист альдостерона. Блокирует обратное
всасывание ионов Na+, уменьшает
таким образом количество жидкости, что приводит к понижению артериального
давления Не влияет на содержание ионов
К+ ! Это очень важно.
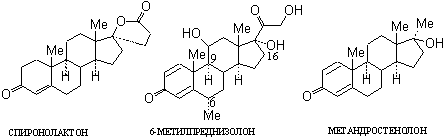
3. Противовоспалительные
средства. Преднизолон. 6-Метилпреднизолон (см. формулу выше). Фторстероиды
(дексаметазон (9a-фтор-16a-метилпреднизолон), триамцинолон (9a-фтор-16a-гидроксипреднизолон. Противовоспалительные мази.
4. Анаболики.
Способствуют образованию мышечной массы и костной ткани. Метандростенолон.
5. БРАССИНОСТЕРОИДЫ
- ПРИРОДНЫЕ СОЕДИНЕНИЯ, ПОМОГАЮЩИЕ РАСТЕНИЯМ БОРОТЬСЯ СО СТРЕССОМ (засуха,
заморозки, излишнее увлажнение), ОБЛАДАЮТ РОСТОРЕГУЛИРУЮЩЕЙ АКТИВНОСТЬЮ.
24-эпибрассинолид
[(22R, 23R,24R)-
2α,3α,22,23- тетрагидрокси-В-гомо-7-окса-5α-эргостан-6-он.
Препарат "Эпин-экстра", ННПП "НЭСТ-М".
МЕТАЛЛОКОМПЛЕКСНЫЙ
КАТАЛИЗ (1 СЕМЕСТР).
Комментарий:
1. Комплексы переходных металлов в
органической химии.
Общие
представления о структуре комплексов (желательно вспомнить 1-й курс, где все
это изучалось). Отличие переходных металлов от непереходных – участие d-электронов в образовании связей с лигандами. Правило
18-ти электронов (аналог правила октета Льюиса в химии переходных металлов).
Металлоорганические
соединения – соединения со связью
металл – углерод (ковалентной или донорно-акцепторной).
Как
считают электроны и формальную степень окисления металла в комплексах:
А)
лиганды – нейтральные молекулы, связь с металлом донорно-акцепторная – лиганд
предоставляет пару электронов, металл – пустую d-орбиталь, металл не изменяет степень окисления.
Примеры таких лигандов – вода, аммиак, амины, пиридин, фосфины, оксид углерода.
Б)
лиганды – одновалентные остатки, связь ковалентная, по одному электрону от
металла и лиганда. Степень окисления металла увеличивается на единицу. Примеры
лигандов – гидроксид, алкоксиды, галогениды, амиды, гидриды, одновалентные
остатки органических соединений (алкилы, ацетилениды, арилы, ацилы и т.п.)
В)
лиганды, связанные с металлом больше чем одним атомом углерода; несколько
примеров: циклопентадиенил, аллил, этилен.
2.
Представление о каталитическом процессе. Важно помнить что а) катализатор
не входит в стехиометрическое уравнение реакции; б) не может изменить положение
равновесия. Если какая-то реакция невозможна по энергетическим причинам,
никакой катализатор не сможет ее сдвинуть с места. Катализатор влияет только на
скорость реакции.
3. Реакции, катализируемые
комплексами палладия и никеля.Pd и Ni близкие аналоги в Периодической системе. Комплексы
катализируют одни и те же реакции по одним и тем же механизмам, но Pd обладает более широкими возможностями, поэтому их
используют гораздо чаще.
Комплексы
нульвалентного палладия – обладают высокой реакционной способностью, но
неустойчивы (разлагаются с образованием металлического палладия), если в координационной
сфере нет прочно связанных лигандов - фосфинов. Комплекс Pd(PPh3)4 устойчив (соответствует правилу 18-ти
электронов – d10 у Pd(0) плюс 4
пары электронов от фосфина).
Реакции
кросс-сочетания. Так называют реакции между электрофилом (RX) и нуклеофилом (R’M) с
образованием продукта сочетания R-R’. Слово "кросс" означает, что части R и R’ как правило,
различны. Остатки R’ и R могут быть практически любыми: но чаще всего
используются ненасыщенные радикалы (арилы, винилы, ацетиленилы, аллилы, бензилы
и т.п.). Вариант кросс-сочетания, где оба радикала алкилы почти не
используется.
RX – любое органическое галогенпроизводное, где X – иод или бром, реже хлор. Также часто используются
трифлаты ROSO2CF3 (см.
тему Нуклеофильное замещение, уходящие группы). Более доступные тозилаты в
кросс-сочетании практически не используются.
R’M –
металлоорганическое соединение, но не любое. В каталитическом кросс-сочетании
используются соединения бора, олова, цинка, магния, кремния
(в порядке убывания значения), очень редко меди.
Производные всех остальных металлов и металлоидов, в том числе и ближайших
аналогов перечисленных, в кросс-сочетании практически не применяются.
4. Механизм кросс-сочетания. В реакции участвуют комплексы палладия или никеля в
степени окисления 0. Реакция включает несколько стадий:
Стадия
0: активация катализатора –
образование координационно-ненасыщенных комплексов Pd(0) за счет диссоциации или восстановления исходных
комплексов. Функция этой стадии – освободить место в координационной сфере
металла для последующих превращений.
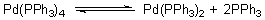
В
собственно каталитический цикл входят три стадии:
Стадия
1. Присоединение электрофила RX к богатому электронами комплексу Pd(0), причем палладий изменяет степень окисления до +2.
Стадия называется "окислительное
присоединение".
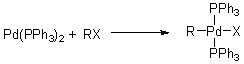
Стадия
2. Образовавшийся комплекс реагирует
с нуклеофильным металлоорганическим соединением, причем происходит замещение
уходящей группы Х на органический остаток. Эту стадию принято называть "переметаллированием".
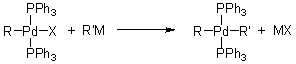
Стадия
3. Образовавшийся комплекс палладия
распадается с образованием искомого продукта R-R’. Палладий при
этом восстанавливается и возвращается к исходной форме. Стадия называется
"восстановительным отщеплением", так как этот процесс, с точки зрения
координационного состояния палладия, обратен к стадии окислительного
присоединения.
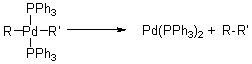
Механизм
кросс-сочетания принято изображать в виде каталитического цикла, на котором
представлены стадии1-3.
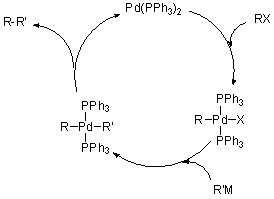
Эффективность
катализатора в конкретных реакциях определяется числом [1]каталитических
циклов, приходящихся на один атом палладия. В большинстве реакций эта величина
составляет от 10 до 1000, но известны реакции, в которых наблюдается до 106-108
циклов.
5. Наиболее важные реакции
кросс-сочетания:
a.
Наиболее часто
используются реакции борорганических соединений с арил- и винилгалогенидами (Реакция
Судзуки-Мияуры)1. Борорганические соединения – бороновые
кислоты и продукты гидроборирования ацетиленов и олефинов. Реакция требует
присутствия основания.
b.
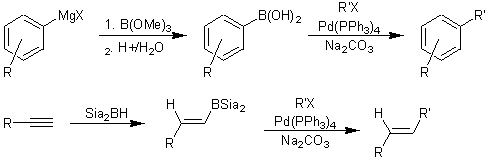
c.
Реакции
оловоорганических соединений (реакция Стилле) и цинкорганических соединений
(реакция Негиши) с арил- и винилгалогенидами. Не требуют добавления оснований.
Катализаторы – соединения Pd(0). Олово- и цинкорганические соединения обычно
получают из галогенидов олова или
цинка, соответственно, и реактивов Гриньяра или литийорганических соединений.
d.
Реакции
магнийорганических соединений (реакция Кумады-Тамао-Корриу). В отличие от предыдущих
случаев используют никелевые катализаторы. Используется практически только для
получения дифенилов.
4)
К реакциям кросс-сочетания относят
также реакцию терминальных ацетиленов с арил и винилгалогенидами (реакция
Соногаширы или, более правильно,
Соногасиры). Здесь нуклеофилом является ацетилен в присутствии основания и
соли меди(I). Реакция широко применяется для
получения разнообразных диарилацетиленов и енинов.
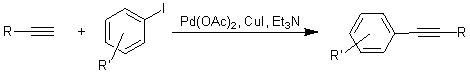
5)
Кроме реакций кросс-сочетания в
органическом синтезе завоевала огромную популярность реакция Мидзороки-Хека
(или просто Хека), формально очень похожая на реакцию Соногасиры, но имеющая
принципиально иной механизм. В реакции Мидзороки-Хека олефины реагируют с
арилгалогенидами или трифлатами. Необходимо добавление оснований (третичных
аминов, ацетата натрия, карбоната калия и т.п.). Наиболее часто в качестве
олефинов используют стирол и производные акриловой кислоты. Продукт образуется
в транс-форме.
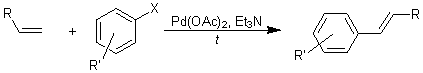
6)
Кросс-сочетание применяется не
только для образования связей углерод-углерод, но и для образования связей С-N, С-O, C-S и других.
Например, в конце 1990-х был разработан наиболее общий метод синтеза аминов –
от первичных до третичных, с любыми органическими остатками
палладий-катализируемым кросс-сочетанием (реакция Бухвальда-Хартвига – см. Амины). Реакция требует применения
комплексов палладия со специальными фосфинами, например,
трис(трет-бутил)фосфином
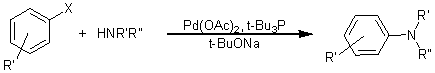
7) Кроме
перечисленных реакций, катализируемых Pd(0), существуют
процессы катализируемые двухвалентным палладием. Pd(2+), в отличие от Pd(0), является электрофилом и окислителем. Реакция с
олефинами протекает аналогично другим реакциям присоединения электрофилов к
олефинам, например, реакции сольвомеркурирования, с той существенной разницей,
что после присоединения электрофила и нуклеофила, происходит самопроизвольное
отщепление гидрида палладия с перегруппировкой енола (рекомендуем вспомнить механизмы реакции сольвомеркурирования олефинов и
гидратации ацетиленов по Кучерову и сравнить).
Регенерация Pd(2+)
осуществляется окислением солями меди и кислородом. В промышленности эта
реакция (ВАКЕР-процесс, по названию компании, впервые запатентовавшей
это превращение) используется как основной метод получения ацетальдегида.
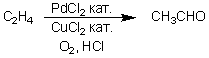
8) Каталитические
превращения органических соединений не исчерпываются химией палладия (и
никеля). Важнейшим каталитическим процессом (Нобелевская премия 2005 года –
Шовен, Шрок, Граббс) стала так называемая реакция метатезиса олефинов. Катализаторами
реакции метатезиса могут быть комплексы как ранних, так и поздних переходным
металлов. Наиболее широко используются катализаторы на основе молибдена и
рутения. Реакция метатезиса протекает через промежуточное образование
комплексов переходных металлов с карбенами.
Два типа катализаторов:
а) карбеновые комплексы Шрока на основе молибдена и
других ранних переходных металлов.
Типичный представитель – компекс Mo(6+) с
карбеном и вспомогательными лигандами. На схемах показывают упрощенно,
показывая только карбен.
б) карбеновые комплексы Граббса на основе Ru(2+). Основной катализатор этого типа кроме
карбенового лиганда имеет вспомогательные лиганды – трициклогексилфосфин и
хлорид. на схемах упрощенно обозначают только карбеновый лиганд. Катализаторы
Граббса наиболее популярны в синтезе.
Реакция метатезиса представляет собой равновесное
превращение.
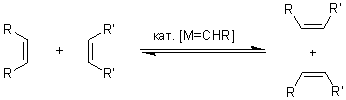
Механизм реакции (Шовен) включает образование
четырехчленного металлацикла и его распад.
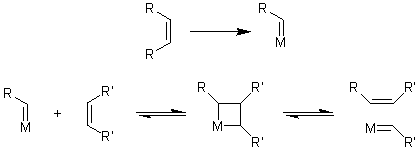
Все стадии обратимы. Чтобы в каждом конкретном случае
понять, в каком направлении пойдет та или иная реакция метатезиса, нужно
оценить относительную устойчивость олефинов (см. тему Алкены) и правило Ле Шателье.
Применение реакции метатезиса:
а) циклизация. Равновесие смещеается за счет удаления
из системы газообразного этилена. Можно получать циклические и гетероциклические
олефины самого разного размера.
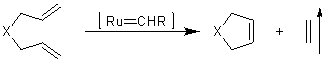
б) Размыкание цикла. Равновесие смещается за счет
избытка этилена под давлением, например
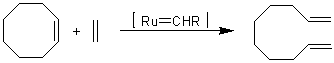
Н.В.Лукашёв, А.В.Чепраков
[1] В отечественной литературе более известна
как реакция Сузуки
|