|
Бутин К.П. Теоретическая стереохимия/ Органическая химия
8.5. Методы разделения энантиомеров
Операции разделения рацемических смесей на
составляющие их оптически активные компоненты
называются расщеплением. Если хотя бы один
энантиомер удается выделить в чистом виде,
расщепление называют полным, в противном случае
говорят, что произошло частичное расщепление,
т.е. оптически активное соединение содержит
примесь второго энантиомера.
Отношение экспериментально наблюдаемого
удельного вращения вещества, полученного путем
расщепления, к удельному (абсолютному вращению
чистого энантиомера называется оптической
чистотой (Р). Тождественными оптической чистоте
являются понятия энантиомерной чистоты или
энантиомерного избытка (э.и.).
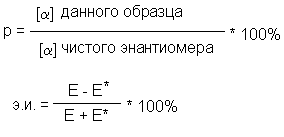
где Е - мольная доля энантиомера, находящегося в
избытке,
Е* - мольная доля другого энантиомера.
Любой процесс получения оптически активного
вещества из оптически неактивного
предшественника, в том числе и расщепление
рацемических смесей, называется оптической
активацией. Общим принципом всех процессов
оптической активации является создание в той или
иной форме диастереомерных взаимодействий.
8.5.1. Расщепление через диастереомеры
Этот метод до настоящего времени использовался
наиболее часто. Если рацемическое соединение
содержит карбоксильную группу, то можно получить
соль с оптически активным основанием. Если же
рацемат содержит аминогруппу, то можно получить
соль с оптически активной кислотой. Допустим, что
оптически активный реагент (в данном случае
основание или кислота) имеет (S)-конфигурацию.
Тогда образующиеся соли будут смесью (R)- и
(S)-диастереомеров, и в отличие от энантиомеров их
свойства будут уже различаться.

В принципе для разделения отличающихся по
свойствам диастереомеров можно использовать
разные методы, но на практике чаще всего
применяют кристаллизацию, т.е. используют
различие в растворимости двух диастереомеров. В
настоящее время все чаще применяют
хроматографические методы. На последней стадии
из соли выделяют знантиомер.
Подбор реагентов для разделения данной
рацемической смеси производится исключительно
эмпирически, т.к. каких-либо теоретических
предпосылок для прогнозирования различной
растворимости диастереомерных солей не
существует.
Для разделения рацемических кислотных
соединений применяют природные оптически
активные основания, которые называются
алкалоидами, например, бруцин, эфедрин, стрихнин,
хинин, цинхонин, морфин и др. После проведения
разделения их регенерируют и используют снова.
Однако эти вещества сильно токсичны и поэтому их
стремятся заменить синтетическими оптически
активными аминами, например, a -фенилэтиламином.
Например, таким путем расщепляется рацемическая
3-метил-2-фенилбутановая кислота.
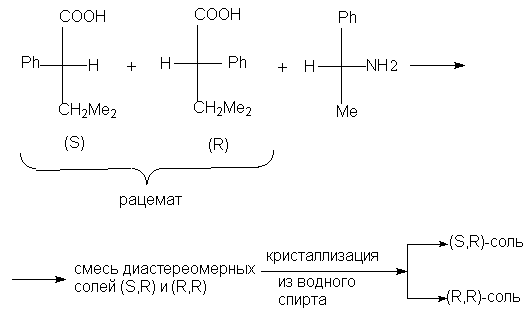
Для разделения рацемических основных
соединений применяют оптически активные
кислоты: винную, миндальную (a
-гидроксифенилуксусную), аспарагиновую
(аминоянтарную), глутаминовую (a
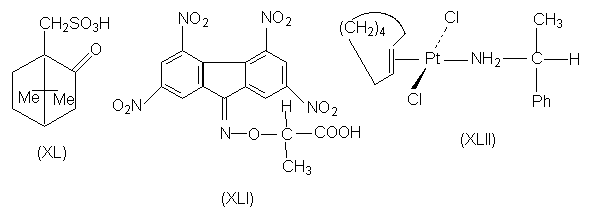
-аминоглутаровую), камфорсульфоновую (XL)и др.
К сожалению, различия в растворимости
энантиомеров редко бывает достаточно велико, для
того чтобы осуществить полное разделение в ходе
одной операции, обычно приходится проводить
многократную кристаллизацию, что делает
разделение длительным трудоемким процессом.
Если молекула не содержит кислотной или
основной группировки, то ее можно сначала ввести,
а затем после разделения на энантиомеры снять,
например,
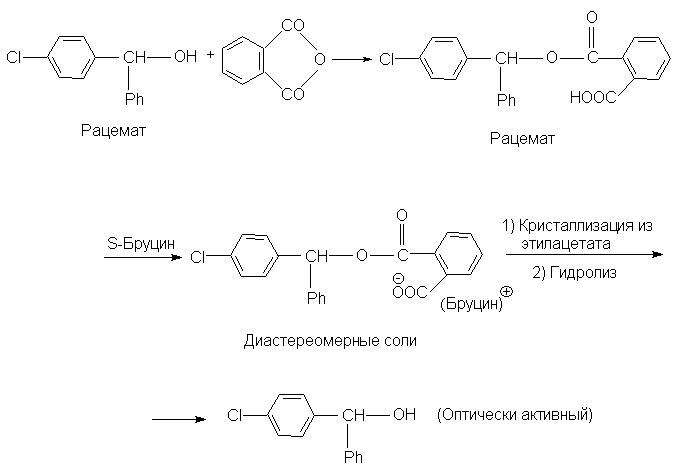
Диастереомеры могут образовываться не только в
результате взаимодействий кислот и оснований
Бренстеда, как описано выше, но также и в
реакциях, в которых взаимодействуют кислоты и
основания Льюиса. Так, при расщеплении
ароматических соединений, в состав которых не
входит ни кислотные, ни основные группировки
(например, хиральных нафтиловых эфиров), может
быть использована их способность образовывать p
-комплексы с нитрофлуореном. Для этой цели
используют реагент (XLI), в котором
элекктроноакцепторные
тетранитрофлуореноноксимная группа придает ей
способность к комплексообразованию с
электронодонорными ароматическими кольцами, а
фрагмент энантиомерной молочной кислоты
обеспечивает реагенту в целом оптическую
активность. Другим примером является
расщепление транс-циклооктена путем
образования комплекса с солью двухвалентной
платины (кислота Льюиса), вторым лигандом у
которой является молекула (R)- a -фенилэтиламина
(XLII).
8.5.2. Хроматографическое расщепление
Если рацемичеcкую смесь хроматографировать на
колонке, заполненной хиральными веществами,
энантиомеры должны проходить с разными
скоростями и, следовательно, их можно разделить.
Таким путем, например, миндальную кислоту
разделяют на колонке, заполненной крахмалом.
Можно использовать бумажную, колоночную, газовую
и жидкостную хроматографию.
8.5.3. Механическое расщепление
В случае рацемической натрийаммониевой соли
винной кислоты энантиомеры при температуре ниже
270 (здесь температура очень важна)
кристаллизуются раздельно: в одном кристалле
собираются (+)-изомеры, а в другом (-)-изомеры. Такие
кристаллы отличаются друг от друга
зеркальностью формы, и их можно разделить с
помощью пинцета и микроскопа. Именно таким путем
Л. Пастер в 1848 г. впервые доказал, что
рацемическая винная кислота в действительности
представляет собой смесь (+)- и (-)-изомеров.
Однако такого рода кристаллизация свойственна
лишь немногим веществам. Описано, например,
расщепление гептагелицена (смесь спирально
сочлененных бензольных колец; аналог
гексагелицена - ). Один из энантиомеров этого
соединения, имеющий необычно высокое оптическое
вращение ([a ]D20= +62000) спонтанно
выкристаллизовывается из бензола.
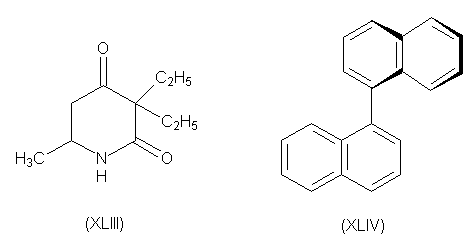
При аналогичном расщеплении
5-метил-3,3-диэтил-2,4-пиперидиндиона (XLIII) было взято
20 кг рацемата и после 400 перекристаллизаций
получено всего 3 г оптически чистого
правовращающего изомера. Одним из немногих
соединений, которые можно разделить пинцетом по
методу Пастера является 1,1,-динафтил (XLIV).
При нагревании рацемата при 76-1500
происходит фазовое изменение с образованием
лево- и правовращающих кристаллов.
8.5.4. Ферментативное расщепление
Довольно часто для получения оптически
активных веществ из рацематов используют
ферменты, которые обладают высокой
стереоспецифичностью действия. Наибольшее
значение метод приобрел для
стереоспецифического гидролиза
N-ациламинокислот. Под действием фермента
ацилазы на рацемическую N- ацетиламинокислоту
L-изомер гидролизуется в 1000 раз быстрее D-изомера,
и после окончания ферментативной реакции легко
можно разделить L-аминокислоту и
D-ацетиламинокислоту.
8.5.5. Установление оптической чистоты
В большинстве случаев при расщеплении
рацематов получаются энантиомеры, не имеющие
100%-ной оптической чистоты. Для установления
содержания в них второго энантиомера применяют
по сути дела те же методы, что и для расщепления, с
той лишь разницей, что в данном случае
образующиеся диастереомерные комплексы не
разделяют, а тем или иным способом определяют их
концентрацию. Относительные концентрации
диастереомеров можно определить любым способом,
например, с помощью ГЖХ или ЯМР-спектроскопии.
8.6. Асимметрический синтез и катализ
Асимметрическим синтезом называют реакции, в
ходе которых один из двух энантиомеров
хирального продукта образуется в большем
количестве, чем второй. В асимметрическом
синтезе ключевой является стадия, в которой так
называемый прохиральный реагент
превращается в хиральный продукт. Прохиральными
называются молекулы, способные превратиться в
хиральные молекулы путем "одношагового"
преобразования структуры. Например,
фенилуксусную кислоту в одну стадию можно
превратить в a -бромфенилуксусную кислоту,
которая хиральна:
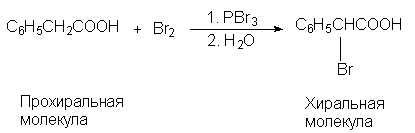
В этой реакции (R)- и (S)-изомерные a
-бромфенилуксусные кислоты образуются в строго
одинаковых количествах (э.и.=0), и, следовательно,
реакцию нельзя назвать асимметрическим
синтезом, хотя она и приводит к образованию
хирального продукта из ахирального. Чтобы
энантиомерный избыток был отличен от нуля,
необходимо обязательно соблюсти одно очень
важное условие. Это условие состоит в том, что в
ходе реакции обязательно должны возникнуть диастереомерые
отношения между вновь возникающим хиральным
элементом (в рассматриваемом примере - центром
хиральности) и вторым хиральным элементом,
специально вводимым в реагирующую систему.
Например, прохиральная молекула натриевой соли
метилэтилмалоновой кислоты при
декарбоксилировании дает рацемическую
2-метилмасляную кислоту, но бруциновая соль дает
продукт с избытком левовращающего изомера:
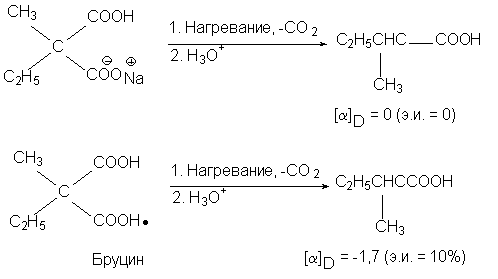
Реакция декарбоксилирования бруциновой соли
явилась первым примером асимметрического
синтеза (В. Марквальд, 1904). Здесь вторым хиральным
элементом является асимметрический центр в
молекуле бруцина.
В том же 1904 г А. Маккензи осуществил
асимметрические синтезы, в которых исходным
реагентом служил (-)-ментиловый эфир
фенилглиоксиновой кислоты (XLV).
В этом примере вторым хиральным элементом
является хиральный центр в ментиловой группе.
Глубокий смысл необходимости присутствия
второго хирального элемента состоит в том, что
Левое будет более предпочтительно, чем Правое
(или Правое более предпочтительно, чем Левое)
лишь в том случае, когда имеется второй элемент,
который тоже может быть левым или правым, и
поэтому "распознает" энергетическую
разницу между подходом реагента (СН3MgI во
втором примере) слева или справа, и способствует
определенной наиболее выгодной ориентации.
В рассмотренных выше примерах вспомогательный
хиральный элемент содержался в самом субстрате.
Однако стереохимический результат реакции
зависит не от симметрии одного лишь реагента, а
от полной симметрии реагирующей системы. Поэтому
при проведении асимметрического синтеза
используют (1) хиральные субстраты, содержащие
прохиральные группы, (2) хиральные реагенты
(например, хиральные гидриды при гидрировании
кратных связей), (3) хиральные катализаторы и (4)
хиральные растворители. Стереоселективность
(энантиомерный избыток) асимметрического
синтеза колеблется в широких пределах, достигая
98% при использовании некоторых хиральных
катализаторов (см. ниже, а также гл. 27), а в
ферментативных реакциях даже 100%. Реакции с
селективностью 100% называются
стереоспецифическими.
Современные представления о механизме
асимметрического синтеза целиком основаны на
конформационном анализе. Но прежде чем перейти к
механизму, необходимо еще раз и более детально
рассмотреть вопросы внутримолекулярной
симметрии, поскольку многие молекулы содержат
атомы или группы атомов, которые лишь кажутся
эквивалентными, но при строгой проверке на самом
деле оказываются разными.
|
