КОМПЬЮТЕРНЫЙ ПРОГНОЗ БИОЛОГИЧЕСКОЙ АКТИВНОСТИ ХИМИЧЕСКИХ СОЕДИНЕНИЙ КАК ОСНОВА ДЛЯ ПОИСКА И ОПТИМИЗАЦИИ БАЗОВЫХ СТРУКТУР НОВЫХ ЛЕКАРСТВ
В.В.Поройков, Д.А.Филимонов
НИИ биомедицинской химии им. В.Н. Ореховича РАМН,
Россия, Москва, 119992, Погодинская ул., 10;
В настоящее время в развитых странах поиск новых лекарств преимущественно основан на скрининге in vitro огромных массивов химических веществ по отношению к сравнительно небольшому числу требуемых видов биологической активности (macromolecular targets). Свойства выявленных таким путем базовых структур (lead compounds) в последующем оптимизируются путем синтеза и исследования большого числа их аналогов. При этом многие виды биологической активности, присущие изучаемым веществам, но являющиеся "побочными" по отношению к избранному направлению исследований, остаются неизученными.
«В наследство» от СССР России, Украине, Казахстану, Армении, Молдове и другим независимым государствам досталась достаточно развитая органическая химия синтетических и природных веществ. Согласно оценкам ведущих фармацевтических фирм, наиболее крупные массивы химических соединений, синтезированных «вручную» (без использования комбинаторной химии) и доступных в настоящее время для скрининга, обеспечиваются химиками из стран СНГ. Возникли и успешно функционируют фирмы-посредники, чей бизнес основан на приобретении у химиков и перепродаже фармацевтическим и агрохимическим компаниям образцов разнообразных веществ для скрининга и заказном синтезе (Specs & BioSpecs, ChemBridge, AsInEx, IBS и др.).
При наличии достаточно богатой коллекции разнообразных химических соединений, страны СНГ обладают крайне ограниченными возможностями для их экспериментального тестирования, что требует тщательнейшего отбора потенциально перспективных веществ уже на ранних стадиях исследования. Такой отбор может быть осуществлен на основе компьютерного прогноза спектра биологической активности химических соединений [http://www.ibmh.msk.su/PASS].
Под спектром биологической активности мы понимаем всю совокупность фармакологических эффектов, биохимических механизмов действия и видов специфической токсичности, которые вещество может проявить при взаимодействии с биологическими объектами. В рамках такого определения мы абстрагируемся от многих факторов, влияющих на количественные характеристики биологической активности (объект, доза, путь введения и т.д.), и рассматриваем биологическую активность как «внутреннее» свойство вещества, которое проявляется при соответствующих условиях в эксперименте или клинике. При этом биологическая активность определяется лишь качественным образом (наличие/отсутствие), что, конечно, является достаточно грубым описанием действительной ситуации, но в рамках такого приближения в аналитических и прогностических целях можно использовать значительный объем информации о биологически активных соединениях, накопленный человечеством.
Целью нашей работы стало создание компьютерной программы, позволяющей прогнозировать большое число вероятных видов биологической активности вещества на основе его структурной формулы с использованием единого описания химической структуры и универсального математического алгоритма установления зависимостей "структура-активность". Программа носит название PASS (Prediction of Activity Spectra for Substances), а ее современная версия (апрель 2002 года) прогнозирует 783 вида биологической активности по структурной формуле химического вещества, включая основные и побочные фармакологические эффекты, механизмы действия, мутагенность, канцерогенность, тератогенность и эмбриотоксичность [http://www.ibmh.msk.su/PASS].
Работа PASS основана на анализе зависимостей «структура-активность» для веществ из обучающей выборки, содержащей более 45000 разнообразных биологически активных веществ (субстанции известных лекарственных препаратов и фармакологически активные соединения). Обучающая выборка постоянно пополняется новой информацией о биологически активных веществах, отбираемой как из публикаций в научно-технической литературе, так и из многочисленных баз данных. Химическая структура представлена в PASS в виде оригинальных MNA дескрипторов (Mulilevel Neighbourhoods of Atoms). MNA дескрипторы имеют универсальный характер и с достаточно хорошей точностью описывают разнообразные зависимости «структура-свойство». Используемый в PASS математический алгоритм был отобран путем целенаправленного анализа и сравнения эффективности для решения подобных задач большого числа различных методов. Показано, что данный алгоритм обеспечивает получение устойчивых в статистическом смысле зависимостей “структура-активность” и, соответственно, результатов прогноза. Это очень важно, поскольку включенные в обучающую выборку данные всегда обладают определенной неполнотой как в отношении охвата всех химических классов веществ, имеющих конкретный вид активности, так и в отношении изученности каждого отдельного вещества на все возможные виды активности.
Средняя точность прогноза при скользящем контроле составляет свыше 85%. Скользящий контроль проводится следующим образом: из обучающей выборки поочередно удаляется одно вещество и для него делается прогноз на основе анализа оставшейся части обучающей выборки, результат сравнивается с известными экспериментальными данными. Процедура повторяется итеративно для каждого из веществ и рассчитывается средняя точность прогноза. Точность прогноза в 85% достаточна для практического применения системы PASS с целью прогноза спектра биологической активности новых веществ, поскольку ожидаемая вероятность случайного угадывания одного из 780 видов активности составляет около 0.1%.
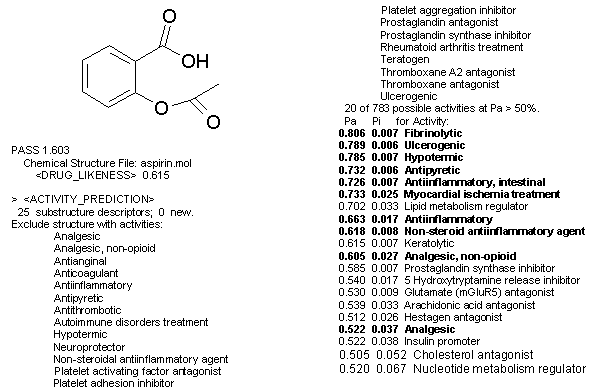
Результаты прогноза выдаются пользователю в виде списка названий вероятных видов активности с расчетными оценками вероятностей наличия (Pa) и отсутствия каждого вида активности (Pi), которые имеют значения от 0 до 1 (рис.1 пример, гиперссылка). Эти вероятности рассчитываются независимо по подвыборкам активных и неактивных соединений, и поэтому их сумма не равна единице. Pa и Pi интерпретируются как оценки меры принадлежности вещества к классам активных и неактивных соединений соответственно, либо как оценки ошибок первого и второго рода. Чем больше для конкретной активности величина Pa и чем меньше величина Pi, тем больше шанс обнаружить данную активность в эксперименте. В дальнейшем мы будем рассматривать ситуации, когда величина Pa достаточно высока и ее значение значительно превосходит Pi. Если при анализе прогнозируемого списка активностей для исследования выбираются те виды активности, для которых Pa>90%, то мы рискуем пропустить около 90% действительно активных соединений, но вероятность ложноположительных прогнозов при этом ничтожно мала; для Pa>80% - пропустим уже только 80% активных соединений, но и вероятность ложноположительных прогнозов будет выше, наконец, для Pa>Pi вероятности ошибок первого и второго рода равны.
На практике, однако, при отборе для исследования наиболее перспективных веществ руководствуются и другими критериями, например, критерием новизны. При этом исходят из того, что чем ближе значение Pa к единице, тем более вероятно, что вещество является близким аналогом известного препарата. Поэтому, если целью исследователя является выявление соединений с достаточно высоким уровнем новизны (New Chemical Entity, NCE), то надо выбирать вещества, для которых величина прогнозируемой вероятности Pa для требуемого вида активности несколько ниже, например, 0,5
Рисунок 1. Химическая структура и прогнозируемый с Pa>50% спектр биологической активности ацетилсалициловой кислоты (жирным шрифтом выделены известные активности; остальные - целесообразно проверить в эксперименте).
Базируясь на данных компьютерного прогноза, исследователь может:
- определить, какие тесты наиболее адекватны для изучения биологической активности конкретного химического соединения.
- обнаружить новые эффекты и механизмы действия для ранее изученных веществ;
- отобрать наиболее вероятные базовые структуры новых лекарств с требуемым биологическим действием среди доступных для скрининга химических соединений.
Система PASS позволяет получить прогноз спектра биологической активности 1000 веществ на обычном персональном компьютере менее чем за одну минуту. Поскольку прогноз выполняется по структурной формуле вещества, он может быть выполнен уже на стадии планирования синтеза.
Применимость PASS для решения практических задач продемонстрирована в многочисленных экспериментах. Прогнозируемые виды активности подтверждены для веществ различных химических классов, проявляющих разнообразные эффекты: противобактериальный, антиаритмический, противоопухолевый, гепатопротекторный, антиамнестический, противоспалительный, антиоксидантный и др. (ссылки на оригинальные публикации - см. на web-сайте [http://www.ibmh.msk.su/PASS]). С применением PASS при поддержке гранта CRDF (RC1-2064) нами был выполнен прогноз спектра биологической активности для 250000 химических соединений, зарегистрированных Национальным институтом рака США (NCI, NIH). Оказалось, что наряду с различными механизмами противоопухолевого действия веществ, для многих соединений предсказываются не известные ранее эффекты. Результаты прогноза представлены на сервере NCI в Интернете [http://cactus.nci.nih.gov/ncidb2/] и применяются для целенаправленного отбора соединений с требуемым спектром биологической активности.
Более подробную информацию о методике компьютерного прогноза биологической активности заинтересованный читатель найдет в Интернете на web-сайте [http://www.ibmh.msk.su/PASS]. Там же реализована возможность прогноза спектра биологической активности веществ через Интернет. Используя стандартные браузеры Nеtscape или Internet Explorer, пользователь может направить на прогноз структурную формулу вещества, представленную в виде mol-файла, (описание – см на: http://www.mdli.com) и автоматически получить на дисплее своего компьютера прогноз видов биологической активности, наиболее вероятных для изучаемого химического соединения. Сотни ученых из многих стран, включая Россию, Украину, Казахстан, США, Германию, Великобританию, и др. уже получили таким образом прогноз биологической активности для нескольких тысяч веществ.
Для поддержания актуального состояния обучающей выборки PASS требуются постоянные усилия по сбору новых данных, их оценке, вводу в компьютер и т.п. Такого рода деятельность применительно к конкретным классам химических соединений и определенным видам биологической активности постоянно проводится каждым исследователем, вовлеченным в процесс создания новых биологически активных веществ. Представляется целесообразным объединение таких исследователей в рамках Ассоциации пользователей PASS. Совместными усилиями, благодаря интеграции имеющейся и новой информации о различных биологически активных веществах, можно было бы создать предпосылки для более надежной компьютерной оценки "биологического потенциала" химических соединений. Более подробная информация об Ассоциации приведена на web-сайте [http://www.ibmh.msk.su/PASS].
Участие в деятельности Ассоциации пользователей PASS обеспечит:
- оснащение программой PASS многих заинтересованных исследователей без уплаты стоимости лицензии за использование программы;
- существенное повышение эффективности исследований, направленных на создание новых биологически активных веществ;
- возможность регулярного получения новых, все более совершенных, версий PASS по мере развития программы.
Дополнительная литература
- Поройков В.В. Компьютерное предсказание биологической активности веществ: пределы возможного. Химия в России, 1999, № 2, 8-12.
- Filimonov D., Poroikov V., Borodina Yu., Gloriozova T. Chemical similarity assessment through multilevel neighborhoods of atoms: definition and comparison with the other descriptors. J. Chem. Inf. Comput. Sci., 1999, 39 (4), 666-670.
- Poroikov V.V., Filimonov D.A., Borodina Yu.V., Lagunin A.A., Kos A. Robustness of biological activity spectra predicting by computer program PASS for non-congeneric sets of chemical compounds. J. Chem. Inform. Comput. Sci., 2000, 40 (6), 1349-1355.
- Lagunin A., Stepanchikova A., Filimonov D., Poroikov V. PASS: prediction of activity spectra for biologically active substances. Bioinformatics, 2000, 16 (8), 747-748.
- Поройков В.В., Филимонов Д.А. Компьютерный прогноз биологической активности химических соединений как основа для поиска и оптимизации базовых структур новых лекарств. В сб.: Азотистые гетероциклы и алкалоиды. Москва: Иридиум-пресс, 2001, т.1, с.123-129.
- Anzali S., Barnickel G., Cezanne B., Krug M., Filimonov D., Poroikov V. Discriminating between drugs and nondrugs by Prediction of Activity Spectra for Substances (PASS). J. Med. Chem., 2001, 4 (15), 2432-2437.
- Poroikov V., Akimov D., Shabelnikova E., Filimonov D. Top 200 medicines: can new actions be discovered through computer-aided prediction? SAR and QSAR in Environmental Research, 2001, 12 (4), 327-344.
- Лагунин А.А., Филимонов Д.А., Поройков В.В. Компьютерный поиск потенциальных антигипертензивных соединений комбинированного действия. Хим.-фарм. журн., 2001, 35 (7), 28-34.
- Poroikov V., Filimonov D. Computer-aided prediction of biological activity spectra. Application for finding and optimization of new leads. Rational Approaches to Drug Design, Eds. H.-D. Holtje, W.Sippl, Prous Science, Barcelona, 2001, p.403-407.
|